





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
三维多孔核壳结构PdNi@Au催化剂的制备及对甲酸的电催化氧化
[李家辉, 秦梦寒, 张洁, 杜仪, 孙冬梅, 唐亚文 ]
]
]
|
|


联系人简介: 唐亚文, 男, 博士, 教授, 博士生导师, 主要从事电化学与能源材料方面的研究. E-mail: tangyawen@njnu.edu.cn
以K2PdCl4/K2Ni(CN)4为前驱体制备了具有凝胶特性的氰胶(Cyanogels), 利用硼氢化钠还原氰胶得到三维多孔珊瑚状PdNi合金前驱体, 在此基础上通过原位Galvanic置换反应, 制备得到内核为PdNi合金、 表面具有不同厚度Au层的三维多孔PdNi@Au催化剂. X射线衍射(XRD)分析和透射电子显微镜(TEM)观测结果显示, 该三维网状结构由粒径约7 nm的纳米颗粒相互连接形成; 能量分散光谱(EDX)线性扫描和元素分布(Mapping)分析显示该催化剂具有典型的核壳结构. 电化学测试结果表明, 表面Au层的厚度影响PdNi@Au催化剂的性能, 当Au的含量(摩尔分数)为5.6%时, 催化剂显示出对甲酸最佳的电催化活性, 对甲酸电催化氧化的峰电流密度达到商业化铂黑催化剂的7.2倍.
K2PdCl4 and K2Ni(CN)4 were employed as precursors to fabricate cyanogel, which was then reduced by NaBH4 to prepare 3D porous coral-like PdNi alloy. On the basis of the synthesized PdNi alloy, 3D porous PdNi@Au catalysts with PdNi alloy as inner core and Au layers of different thickness on the surface were synthesized by in situ Galvanic replacement between PdNi alloy and HAuCl4 aqueous solution. X-Ray diffraction(XRD) and transmission electron microscopy(TEM) indicated that the 3D network structure was composed of interconnected nanoparticles with diameter of 7 nm. Energy dispersive X-ray(EDX) line scanning and mapping could declare its typical core-shell structure. Electrochemical measurements demonstrated that the electro-catalytic performance of PdNi@Au catalysts could be affected by the thickness of Au layer. When the content of Au reached a value of 5.6%(molar fraction), PdNi@Au catalyst exhibited the best catalytic performance for formic acid electro-oxidation. In this case, the peak current density of PdNi@Au catalyst was 7.2 times that of commercial Pd black.
金(Au)基纳米材料因独特的物理化学特性而在催化、 传感、 医疗诊断及光学领域具有潜在的应用价值[1~5]. 其形貌、 尺寸和晶体结构对其物理化学性质影响显著. 处于纳米尺度的Au显示出特殊的物理化学性质, 因此, 许多研究者致力于Au基纳米材料的可控合成[6, 7]. 迄今, 不同维度和形状的Au纳米材料已被合成出来, 如一维的Au纳米线[8, 9]、 纳米棒[10, 11]; 二维的纳米片[12]、 纳米带[13, 14]; 三维的纳米珊瑚[3, 15]、 纳米花[16, 17]、 纳米星[18]、 树状纳米晶[19]等. 其中, 三维多孔Au基材料由于含有较多的孔洞和孔隙率、 较大的比表面积、 丰富的缺陷原子和稳定的互联结构, 使其兼具结构材料和功能材料特点, 其合成方法也受到了研究者们越来越多的关注[20, 21].
目前, 三维多孔Au基纳米材料的合成方法有模板法[22]、 去合金法[23]及电化学方法[24]等. Xu等[25]通过将AgAu合金浸渍在一定浓度硝酸中除去合金中的Ag制备了纳米多孔金, 并将其应用于CO的低温氧化. Liu等[26]发现, 超细网状金纳米线在一定实验条件下能发生自组装进而生成纳米多孔金薄膜, 并将其用于单分子的检测. Kafi等[27]通过简单的水热法在Ti基底上直接生长了三维多孔的Au纳米网络, 其多孔结构能够提供较大的比表面积, 因而成为生物大分子的良好载体.
研究发现, 由于Au和Pd原子之间的电子耦合和协同效应, Au的引入可以提高Pd对甲酸的电催化氧化活性和稳定性[28, 29, 30]. 本文通过氰胶路径制备得到三维网状结构的PdNi合金, 并通过原位Galvanic置换反应制备出了外层具有不同厚度Au的三维多孔核壳结构催化剂(PdNi@Au), 研究了该催化剂对甲酸的电催化氧化性能.
氯亚钯酸钾及氯金酸购于上海笛柏化学品技术有限公司; 镍氰化钾和Nafion溶液购于美国Sigma Aldrich公司; 甲酸购于西陇科学股份有限公司; 钯黑购于上海河森电器有限公司; 硫酸购于国药集团化学试剂有限公司; 硼氢化钠购于上海阿拉丁生物科技股份有限公司. 所用试剂均为分析纯.
D/max-Rc型X射线衍射仪(日本理学公司, Cu Kα 射线, λ =0.154056 nm, 测试电压40 kV, 测试电流100 mA); 场发射电子扫描显微镜(SEM, Hitachi S5500, 日本日立公司); JEM-2100F型透射电子显微镜(TEM, 日本电子株式会社, 加速电压为200 kV); X射线光电子能谱仪(XPS, ESCALAB 250, 美国Thermo VG Scientific公司, 真空度为10-6 Pa, 结合能以C1s 284.6 eV为标准进行校正).
在超声条件下, 向2 mL 0.05 mol/L K2PdCl4溶液中加入1 mL 0.05 mol/L K2Ni(CN)4溶液, 室温静置5 min, 形成黄色果冻状氰胶后滴加6 mL 0.05 g/mL的NaBH4溶液. 室温反应6 h后离心洗涤, 于40 ℃真空干燥, 即制得三维网状PdNi合金. 称取PdNi合金10 mg, 加入20 mL水, 超声, 分别加入0.05, 0.1, 0.2和0.3 mL 0.0486 mol/L的HAuCl4, 于30 ℃反应6 h后离心洗涤, 于40 ℃真空干燥. 4种产物中Au的含量(摩尔分数)分别为2.8%, 5.6%, 11.2%和16.8%, 分别命名为PdNi@Au-2.8, PdNi@Au-5.6, PdNi@Au-11.2以及PdNi@Au-16.8.
电化学测试采用传统的三电极体系, 以Pt丝为辅助电极, 饱和甘汞电极(SCE)为参比电极. 工作电极制备如下: 将10 mg催化剂加入5 mL 去离子水中, 超声30 min. 移取6 μ L悬浊液, 滴涂至直径为3 mm的玻碳电极表面, 室温干燥后, 在催化剂层上滴涂2 μ L 5.0%(质量分数)的Nafion溶液, 再次干燥后即得到工作电极. 电化学测试前向溶液中通15 min氮气以去除溶解氧. 电化学测试在CHI 760D电化学工作站(上海辰华仪器有限公司)上进行, 扫描速率为50 mV/s, 测试温度为(30± 1) ℃.
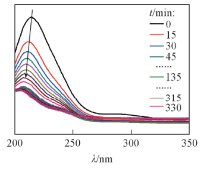
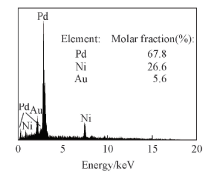
Scheme 1是PdNi@Au催化剂的合成示意图. K2PdCl4和K2Ni(CN)4能够在常温下聚合形成氰胶(一种具有凝胶特性的黄色双金属无机高分子聚合物)[Scheme 1(A)]. 结构示意图显示出氰胶中的金属物种集中在三维骨架上, 还原生成的金属核沿氰胶骨架相互连接而形成三维结构的PdNi纳米聚集体[Scheme 1(B)]. 利用该三维纳米聚集体作为前驱体, 加入HAuCl4后, Au(Ⅲ )与PdNi合金之间发生Galvanic置换反应, 形成内层为PdNi合金, 表面为Au的三维多孔核壳结构[(PdNi@Au, Scheme 1(C)]. 由于3种金属的金属活动性顺序为Ni> Pd> Au, 当向PdNi合金中加入HAuCl4时, Au(Ⅲ )会首先置换Ni原子, 当Ni原子反应完后, Au(Ⅲ )会继续置换Pd原子. 紫外光谱(图1)显示, 随着反应时间的延长, HAuCl4在230 nm处的特征吸收峰逐渐降低, 当反应进行135 min后, HAuCl4的特征吸收峰强度不再变化, 显示出Au(Ⅲ )已经完全被取代. PdNi@Au-5.6催化剂的EDX测试结果(图2)表明, 催化剂中Au的含量与理论值相符.
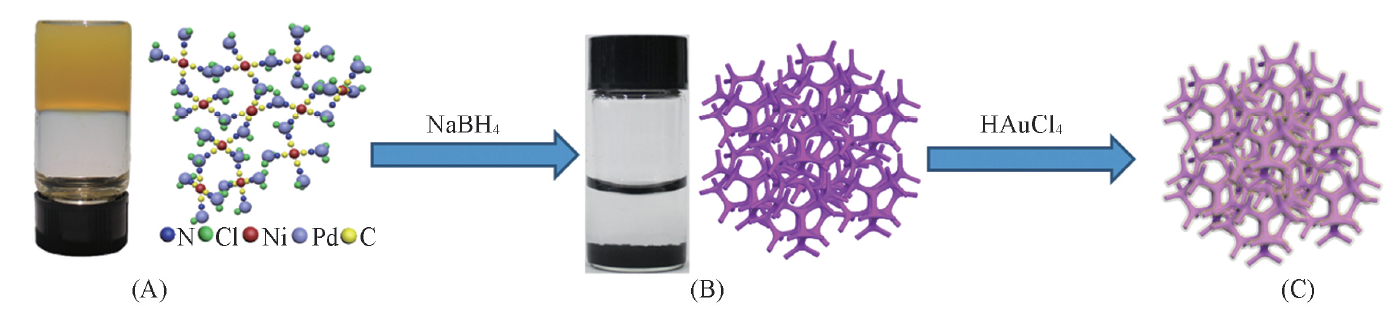 | Scheme 1 Synthetic routes of PdNi@Au catalyst (A) Orange jelly-like PdNi cyanogel and its ball-stick model; (B) photograph of 3D porous PdNi alloy with network structure; (C) schematic diagram of PdNi@Au catalyst. |
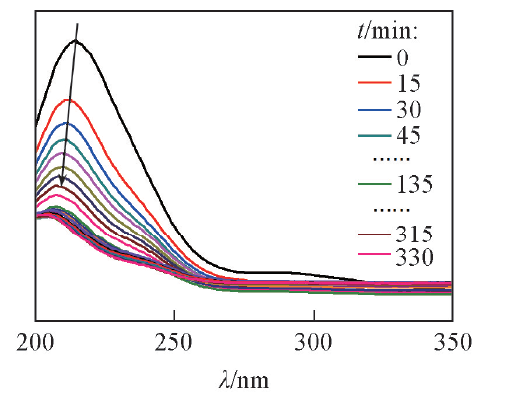 | Fig.1 UV spectroscopy of reaction system at different time |
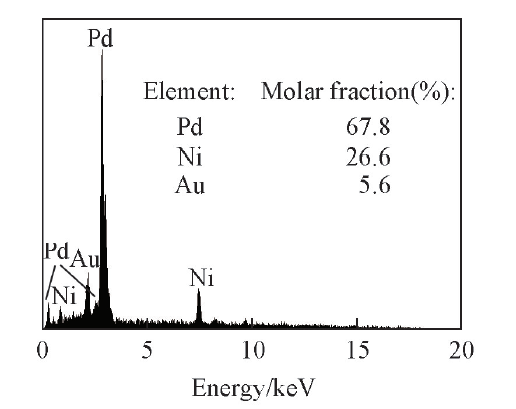 | Fig.2 EDX pattern of PdNi@Au-5.6 catalyst(B) |
图3(A)为PdNi@Au-5.6催化剂的SEM照片. 可以看出, PdNi@Au-5.6催化剂的形貌是具有多级孔洞的珊瑚状结构, 非常有利于燃料分子的传输、 CO2分子的释放及电子的传递[31, 32]. 图3(B)和(C)为PdNi@Au-5.6的TEM照片. 可看出三维网状结构中存在许多大小不一的孔洞[图3(B)中红色虚线标出处], 与SEM照片一致. 选区电子衍射[SAED, 图3(B)插图]证明PdNi@Au-5.6催化剂为多晶结构. HRTEM[图3(C)]进一步显示, PdNi@Au-5.6催化剂的三维网状结构是由粒径约7 nm的金属纳米粒子构成, 由于Au覆盖在纳米粒子的表面, 因此很容易观察到Au的晶格条纹,
 | Fig.3 Typical SEM(A), TEM(B) and HRTEM(C) images of PdNi@Au-5.6 catalyst The insets in (B) and (C) are corresponding SAED pattern and the outer Au layer, respectively. |
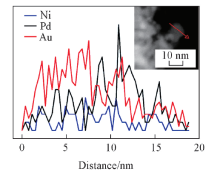
同时, 在局部也能观察到Pd的晶格条纹, 这一方面证明了PdNi@Au-5.6催化剂具有核壳结构, 同时也表明Au层在Pd金属外面的包裹非常薄, 其厚度为12 nm[图3(C)插图]. 能量色散光谱(EDX)线性扫描(图4)也证明PdNi@Au-5.6催化剂是外部富Au, 内部富Pd, Ni的核壳结构. HAADF-STEM照片[图5(A)]表明此材料呈多孔珊瑚结构, 与SEM及HRTEM表征结果一致. EDX-mapping图[图5(B)(E)]中, Au元素的分布面积最大, 镍元素的分布面积最小, 这进一步证明了PdNi@Au-5.6催化剂的核壳结构.
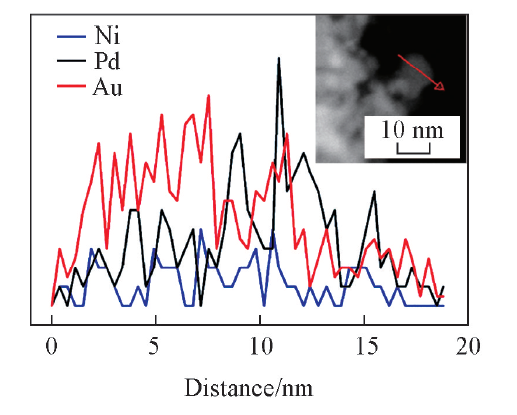 | Fig.4 EDX line scanning pattern of PdNi@Au-5.6 catalyst Inset is corresponding HRTEM image of the EDX line scanning test region. |
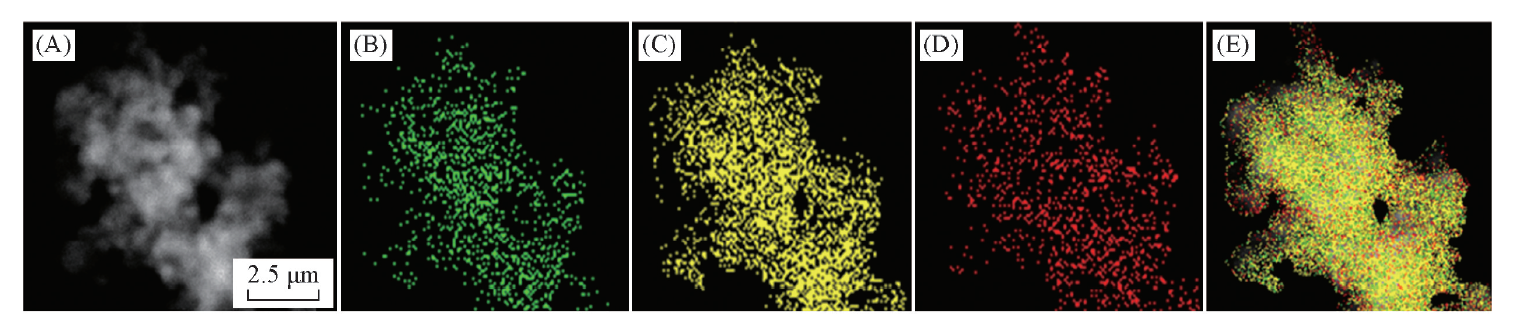 | Fig.5 HAADF-STEM image(A) and EDX mapping patterns of PdNi@Au-5.6 catalyst(B— E) (B) Ni; (C) Pd; (D) Au; (E) overlap. |
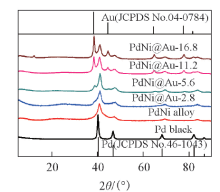
图6为催化剂的XRD谱图. 6种催化剂均为面心立方(fcc)晶体结构. 与纯Pd的标准卡片(JCPDSNo.46-1043)对比可以看出, Ni的引入导致Pd的衍射峰位置正移, 这证明了PdNi合金的生成. 随着Au含量的增加, PdNi@Au-2.8, PdNi@Au-5.6, PdNi@Au-11.2和PdNi@Au-16.8催化剂中Au的衍射峰强度逐渐增强, 表明催化剂表层Au的厚度不断增加.
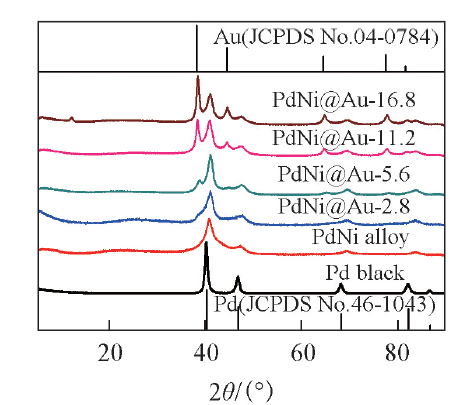 | Fig.6 XRD patterns of different catalysts |
PdNi@Au-5.6催化剂的XPS谱图[图7(A)]显示样品中有Pd, Ni, Au 3种元素存在, 与EDX结果一致. 图7(B)和(C)分别为Pd和Au在P
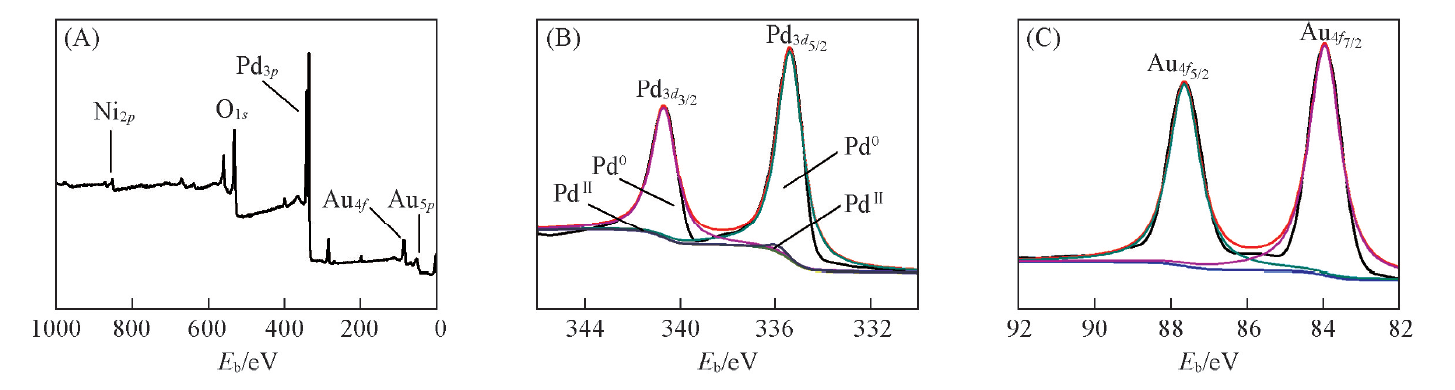 | Fig.7 XPS spectra of PdNi@Au-5.6 (A) Full scan; (B) Pd3d region; (D) Au4f region. |
图8(A)是催化剂在氮气饱和的0.5 mol/L H2SO4中的循环伏安(CV)曲线. 可以看到, 正向扫描时在0.450.96 V范围内出现的峰是Pd的氧化峰, 反向扫描时Pd的氧化物/氢氧化物在0.5 V 附近被还原. 由于Pd对氢有吸附作用, 只能通过积分Pd氧化物的还原峰来计算电化学活性面积(ECSA). 计算结果表明, PdNi@Au-5.6催化剂的ECSA为30.92 m2/g PdAu, 与PdNi合金催化剂(30.43 m2/g Pd)接近, 约为Pd黑催化剂(10.37 m2/g Pd)的3倍, 这可能是由于前二者均具有三维多孔珊瑚状结构所致.
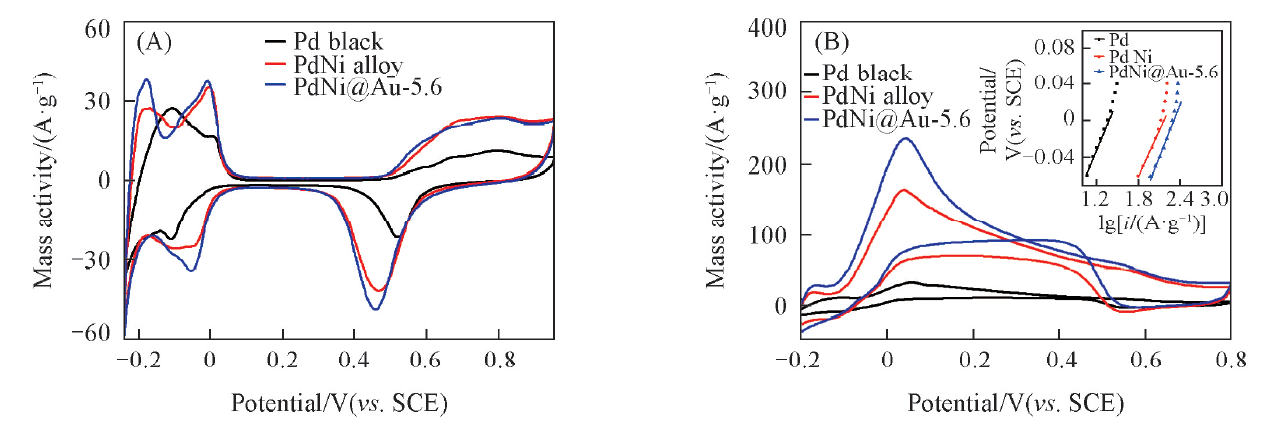 | Fig.8 CV curves of different catalysts in N2-saturated 0.5 mol/L H2SO4(A) and in N2-saturated 0.5 mol/L H2SO4+0.1 mol/L HCOOH(B) Inset in (B) is corresponding Tafel plots. |
图8(B)是催化剂在氮气饱和的0.5 mol/L H2SO4 + 0.1 mol/L HCOOH中的CV曲线. 甲酸在PdNi@Au-5.6催化剂上氧化的峰电流达到了235.62 A/g PdAu, 分别是PdNi合金和Pd黑的1.4倍(164.16 A/g Pd)和7.2倍(32.69 A/g Pd), 这可能是由于Au掺杂导致Pd电子结构改变造成的, Hu等[34]的研究也证明了这一点. Tafel曲线[图8(B)插图]显示, 在同样的输出电流密度下, PdNi@Au-5.6催化剂极化过电位最小, 这表明Au能促进甲酸在Pd催化剂上氧化的动力学过程. 图9(A)为催化剂的计时电流曲线. 可以看出, 与PdNi合金和Pd黑催化剂相比, PdNi@Au-5.6拥有更高的催化活性和稳定性, 这可能是由于三维网状的多孔结构提供了更多的活性位点, 提升了催化剂的催化活性; 同时Au的引入促进了催化剂表面活性含氧基团的生成, 因而提高了催化剂的稳定性[35]. CO溶出伏安测试[图9(B)]显示, CO在PdNi@Au-5.6催化剂上的起始氧化电位与PdNi和Pd黑相比分别负移了56和104 mV, 峰电位分别负移了37和44 mV, 这说明PdNi@Au-5.6催化剂能在更低的电位下氧化CO, 具有更好的抗毒化性能.
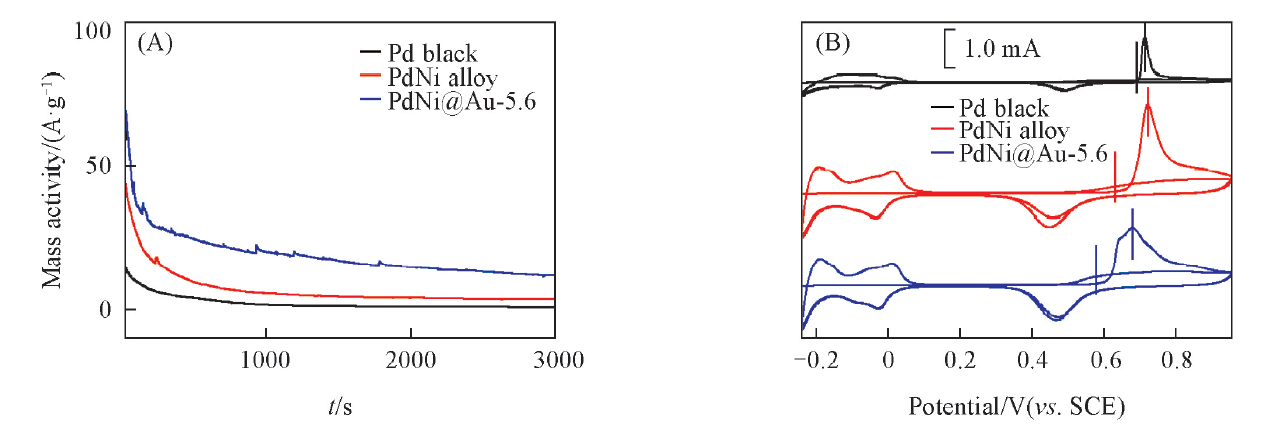 | Fig.9 Chronoamperometry curves at 0 V for 3000 s in N2-saturated 0.5 mol/L H2SO4+0.1 mol/L HCOOH(A) and CO-stripping voltammograms in N2-saturated 0.5 mol/L H2SO4(B) of different catalysts |
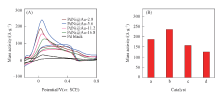
Au含量对PdNi@Au催化剂性能的影响如图10所示. 可见, 随着HAuCl4的加入, 催化剂对甲酸氧化的电催化性能明显提升, 这说明Au能够提升Pd金属催化剂对甲酸氧化的电催化性能. 理论上, 单原子层的金能得到最佳的电子相互作用及最佳的催化性能, 然而, 实际制备很难得到均一分布的单层Au原子.
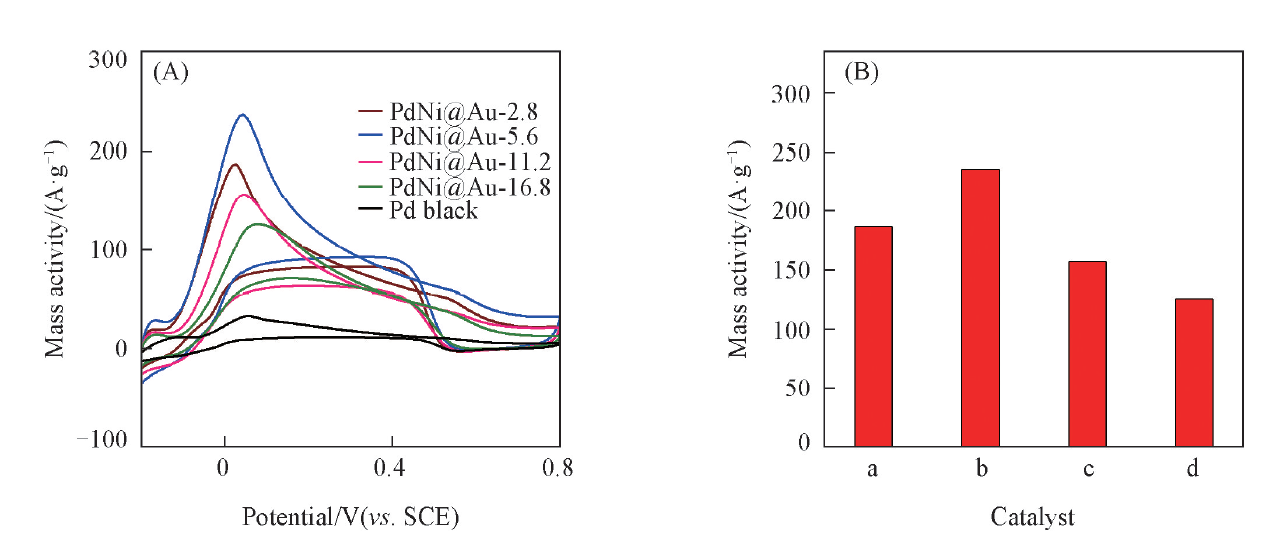 | Fig.10 CV curves of different catalysts in N2-saturated 0.5 mol/L H2SO4+ 0.1 mol/L HCOOH solution(A) and histogram of mass-normalized peak current for different catalysts(B) a. PdNi@Au-2.8; b. PdNi@Au-5.6; c. PdNi@Au-11.2; d. PdNi@Au-16.8. |
研究结果显示, 当催化剂中Au的含量为5.6%, 表面Au层厚度为1~2 nm时, PdNi@Au性能最佳, 其催化甲酸氧化的峰电流密度达到235.62 A/g PdAu, 然而当Au含量进一步增加时, Pd表面的Au层厚度随之增加, 这可能会封闭Pd的表面活性位点, 抑制甲酸在Pd表面的吸附, 因此加入过量的Au反而降低了Pd对甲酸的电催化氧化活性.
通过还原K2PdCl4/K2Ni(CN)4氰胶制备了三维多孔珊瑚状PdNi前驱体, 在此基础上通过原位Galvanic置换反应制备得到一系列不同Au含量的PdNi@Au核壳催化剂. 电化学测试结果表明, Au能够有效促进Pd金属催化剂对甲酸的电催化氧化活性和稳定性; 当Pd金属催化剂表面Au层厚度为12 nm时, 催化剂性能最佳.
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|
| [20] |
|
| [21] |
|
| [22] |
|
| [23] |
|
| [24] |
|
| [25] |
|
| [26] |
|
| [27] |
|
| [28] |
|
| [29] |
|
| [30] |
|
| [31] |
|
| [32] |
|
| [33] |
|
| [34] |
|
| [35] |
|