
{kind=link}
{kind=link}
{kind=link}
{kind=link}
卟啉-碳硼烷-硼亚甲基二吡咯三元化合物二阶非线性光学性质的理论研究
[康慧敏, 王洪强, 王慧莹, 吴黎歆, 仇永清 ]
]
]
|
|


联系人简介: 仇永清, 男, 博士, 教授, 博士生导师, 主要从事应用量子化学研究. E-mail: qiuyq466@nenu.edu.cn
采用密度泛函理论(DFT)方法对卟啉-碳硼烷-硼亚甲基二吡咯(BODIPY)三元化合物的几何结构、 吸收光谱及二阶非线性光学(NLO)特性进行计算分析. 结果表明, V型化合物的静态第一超极化率( βtot)大于相应直线型化合物, 且延长共轭链可提高体系的 βtot. 分析体系的电子密度差分图得出, 化合物氧化还原态的电荷转移方式与本征态相比发生了改变, 从而使其二阶NLO性质发生明显变化. 含频第一超极化率计算结果表明, 在一定范围内频率对化合物有较小的色散效应. 因此, 通过延长二维化合物的共轭链及氧化还原反应, 可以有效调控其二阶NLO响应.
The calculations of geometric structures, electronic absorption spectra, second-order nonlinear optical(NLO) properties of porphyrin- o-carborane-boron-dipyrromethene(BODIPY) triad were carried out by density functional theory(DFT). The results show that the static first hyperpolarizability( βtot) of the V-shaped compound is larger than that of the linear-shaped compound, and the βtot values can be enhanced by extending π-conjugate bridge. The analysis of electron density difference maps can be seen that the charge transfer pattern of the oxidized/reduced species have changed, which lead to the second-order NLO properties have significantly varied. The investigation of the frequency-dependent first hyperpolarizability shown that less dispersion effect at restrictive frequency region for all of the compounds. Therefore, second-order NLO properties can be effectively modulated by extending π-conjugate bridge and oxidized/reduced reactions.
近年来, 随着光通信、 光信息处理的深入研究及广泛应用, 非线性光学(NLO)材料得到了迅速发展[1~5], 在NLO性质研究中, 材料的二阶NLO效应日益受到重视, 其相关应用也最为广泛[6, 7, 8, 9], 常应于激光组件的开发、 光学信息处理和数据存储及光电器件的制造. 常见的二阶NLO材料包括无机材料、 有机材料、 聚合物以及有机金属配合物等[10~12]. 相比无机材料, 有机材料具有较低的介电常数及更为灵活的设计特性, 受到许多理论与实验研究者的青睐[13]. 量子化学计算方法通过对分子进行合理的设计和优化、 分析分子结构与NLO性质的关系, 为NLO材料的制备提供了有力的理论依据[14, 15].
卟啉类NLO材料由于响应速度快、 灵活性好和易于修改等特性引起了人们的关注, 这类材料常应用于光电技术、 信号处理及超快光通信等方面[16, 17, 18, 19], 卟啉中高度可离域的π 电子对提高其NLO响应起到了重要作用. 硼亚甲基二吡咯(BODIPY)衍生物具有良好的光学特性和光学可调性, 在生物成像和传感探头等医学领域有重要的应用, 随着BODIPY衍生物的不断合成, 对其NLO性质的研究也有较多报道[20, 21, 22]. 碳硼烷通过顶点的C成键, 形成明显的非对称电荷分布特征及强的吸电子性, 具有刚性的笼状结构和独特成键特征以及良好的化学稳定性, 引起研究者对其NLO性质的研究[23, 24].
2017年, Berksun等[25]合成了直线型卟啉-乙炔-BODIPY(1)和V型卟啉-o-碳硼烷-BODIPY三元化合物(3), 并研究了其紫外光谱、 核磁共振谱及电化学性质等, 表明卟啉-BODIPY为典型的D-A结构, 有明显的电荷转移. 目前, 对卟啉类[26]、 BODIPY类[27]和碳硼烷类[28]化合物均有相关NLO性质的计算, 但卟啉-碳硼烷-BODIPY三元化合物二阶NLO性质未见报道, 本文在化合物3的基础上设计了直线型卟啉-p-碳硼烷-BODIPY异构体2, 并通过延长化合物3中共轭π 桥设计了化合物4(图1). 采用密度泛函理论(DFT)方法对这些化合物及其氧化还原态的几何结构、 自然键轨道(NBO)电荷、 吸收光谱和二阶NLO性质等进行计算分析, 讨论了碳硼烷、 体系构型、 共轭π 桥及氧化还原过程对其二阶NLO性质的影响, 为设计合成新型NLO材料提供了一定的理论指导.
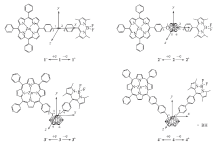
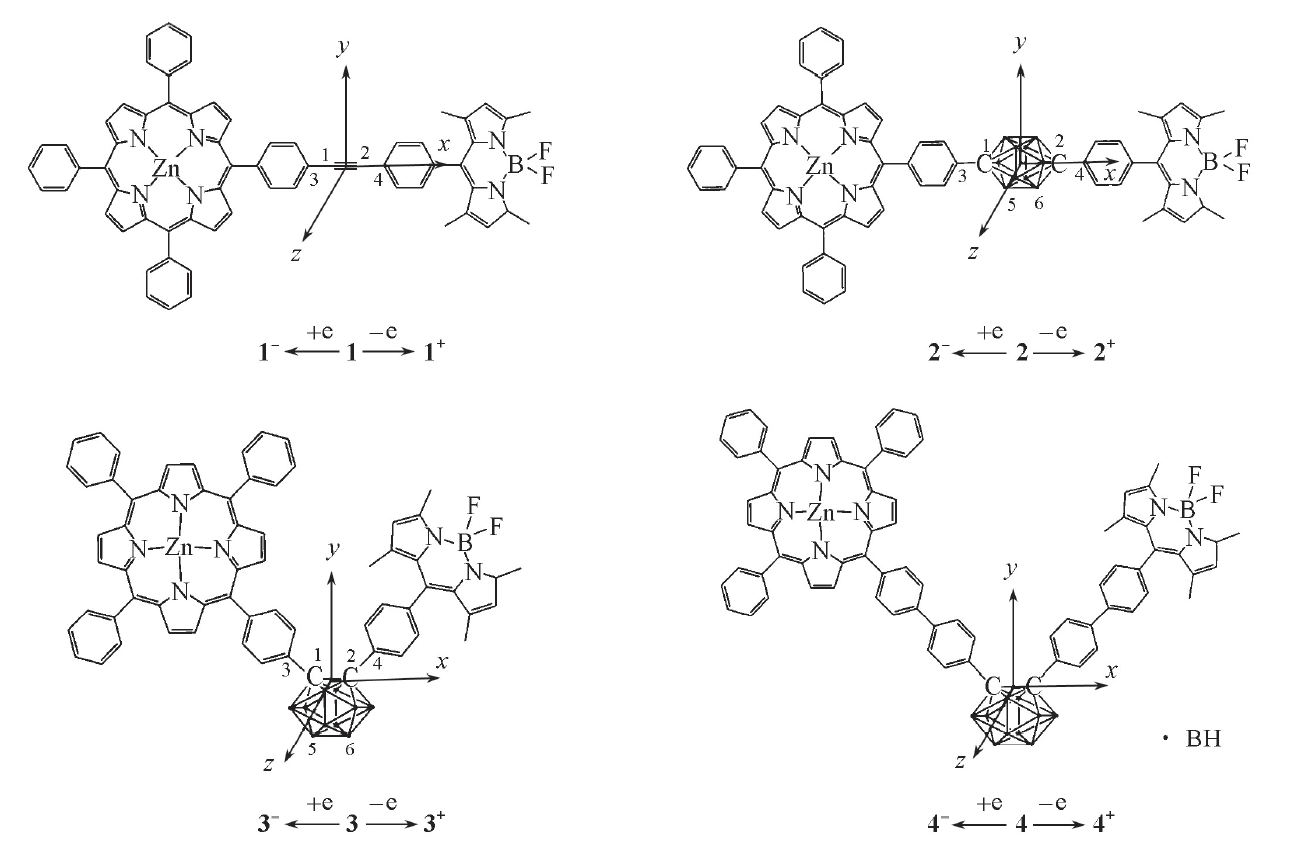 | Fig.1 Geometry structures of compounds 1— 4 |
采用B3LYP方法对所有化合物几何构型进行优化并进行频率计算, 所有化合物均无虚频, 表明得到了稳定几何构型. 采用与优化几何构型相同的方法计算化合物的NBO电荷, 采用ω B97XD和M06-2X 2种方法结合解析导数法计算化合物的第一超极化率. 所有计算中, 对非金属原子采用6-31G* 极化基组, 对金属原子Zn采用LANL2DZ有效核势基组.
在均匀的电场中, 分子的能量可按Taylor级数展开[29]:
式中: 各下标取笛卡尔坐标x, y和z; E(0)为无外场时分子的能量; μ i为分子偶极矩向量的分量; Fi为外电场在i方向分量; α ij为线性极化率张量; β ijk和γ ijkl分别为分子二阶和三阶非线性极化率张量. 分子第一超极化率总有效值β tot(a.u.)计算如下:
实验测量第一超极化率值是在一定频率的激光场中, 为了使理论计算结果更好地模拟实验, 采用耦合微扰密度泛函理论(CP-DFT)计算体系的含频第一超极化率[β (ω )(a.u.)]. β (ω )由下式计算:
为了进一步解释分子的二阶NLO响应, 采用含时密度泛函理论(TD-DFT) B3LYP方法模拟了分子的吸收光谱. 为了得到准确的激发能, 计算过程中考虑了溶剂化效应(PCM)[30, 31](以甲苯作为溶剂模拟实验条件). 所有计算均采用Gaussian 09W[32]程序包完成.
化合物1和3已被实验合成, 且化合物3的晶体数据已有报道[25]. 化合物14及其氧化态和还原态化合物优化的部分几何参数列于表1. 由表1可知, B3LYP方法和6-31G* /LANL2DZ基组优化得到的构型与实验值相吻合, 说明所选用的方法和基组适合本文体系.
| Table 1 Partial bond lengths of the studied compounds as obtained using DFT(B3LYP functional) employing the 6-31G* /LANL2DZ basis set |
化合物2的C1— C3和C2— C4键长比化合物1的增加约0.01 nm, 这与碳硼烷的非共轭性相关, 其它键长变化较小. 化合物2的C1— C3和C2— C4键长比化合物3的略有增加, 说明对位碳硼烷中碳原子的成键能力略小于邻位碳硼烷, 其它键长几乎无变化. 化合物4的C1— C2键长相对于化合物3增加0.0011 nm, 其它键长变化较小, 可见共轭链的增长对键长影响较小. 相对于本征态, 氧化态化合物1+4+中C1— C2, C1— C3和C2— C4键长几乎无变化, Zn— Nmean键长约增大0.0005~0.0007 nm, B— N键长约增加0.0002~0.0004 nm, 而B— F键长减小约0.0003~0.0005 nm. 还原态3-和4-的C1— C2键长分别增加0.0595和0.0566 nm, 说明还原后邻位碳硼烷发生扩张; 对于C1— C3和C2— C4键长, 在化合物1-~4-中都有所减小, 说明还原体系中C1和C2的成键能力增强. 以上分析表明, 氧化还原对化合物几何构型有一定影响, 可能会影响其二阶NLO性质.
化合物1~4选取的坐标系如图1所示. 选择可靠的方法计算第一超极化率是非常重要的, 通常认为B3LYP方法结果高估了β 值, 为验证计算结果的可靠性, 选择长程校正ω B97XD和M06-2X 2种方法对β tot进行计算, 所得结果的大小顺序均为β tot(2)< β tot(1)< β tot(3)< β tot(4)(表2). 为方便讨论, 选择ω B97XD方法计算的结果进行对比分析.
| Table 2 Computed static first hyperpolarizability for compounds 1— 4 employing the 6-31G* /LANL2DZ basis set |
由于化合物1和2为直线型结构, 对β tot的贡献主要为β x分量, 且β x为负值, 表明体系电荷转移方向为沿x轴正向. 由于化合物3和4为V型结构, β tot值来源于β x和β y的共同贡献, β x为负值, β y为正值, 表明电荷转移方向沿x轴正向和y轴负夹角方向. 由表2可见, 化合物2的β tot值小于化合物1, 源于化合物1的乙炔桥良好的共轭性有利于电荷转移; 化合物3的β tot值是化合物2的18.6倍, 可见V型分子由于电荷转移形式的变化比直线型分子具有更大的二阶NLO系数. 延长共轭桥后, 化合物4的β tot值为化合物3的1.4倍, 因此延长共轭桥也是增大二阶NLO系数的一种方式.
为深入理解化合物结构与二阶NLO响应之间的关系, 采用TD-B3LYP, TD-CAM-B3LYP及TD-M06L等方法在6-31G* /LANL2DZ基组水平上模拟了化合物的吸收光谱, 其中B3LYP方法模拟结果与实验值[25]较吻合.
讨论了化合物的主要激发态的电子跃迁性质, 跃迁参数列于表3, fos和Egm分别表示化合物的振子强度和跃迁能. 绘制了吸收光谱图及主要激发态的电子密度差分图(EDDM)(图2), 电子密度差的计算如下:
| Table 3 Detailed TD-DFT calculations for compounds 1— 4 employing the 6-31G* /LANL2DZ basis set |
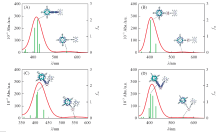
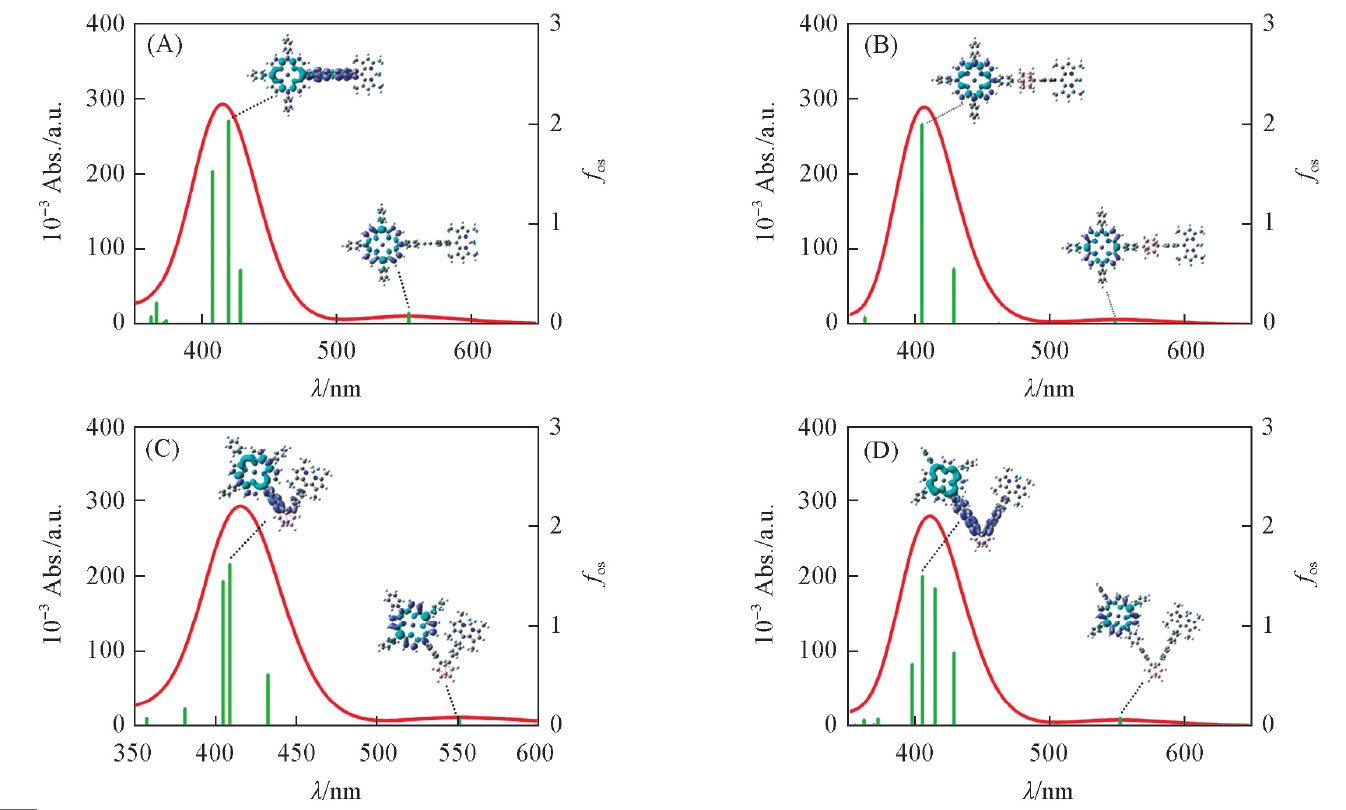 | Fig.2 UV-Vis spectra of the compounds 1(A), 2(B), 3(C) and 4(D) along with electron density difference maps(EDDM) of corresponding transitions Purple and blue colors indicate accumulation and depletion of electron density, respectively. |
式中: ρ ele(r)和ρ hole(r)分别表示电子和空穴分布.
由图2可知, 化合物1的吸收光谱包含一个低能吸收峰(553.6 nm)和一个高能吸收峰(420.0 nm), 结合电子密度差分图(EDDM)可知, 高能吸收峰表现为卟啉向乙炔基苯的电荷转移, 低能吸收峰表现为卟啉局部电子激发(图S1, 见本文支持信息). 化合物2包含一个位于548.7 nm处的低能吸收峰和一个位于404.9 nm处的高能吸收峰, 跃迁形式均为卟啉局部电子激发, 与化合物1相比, 化合物2电荷转移只发生在卟啉内部, 导致其较弱的二阶NLO效应. 化合物3包含一个位于408.8 nm处的主要吸收峰以及一个位于551.4 nm处的低能吸收峰, 其跃迁形式为卟啉向π 桥的电荷转移及卟啉局部电子激发. 此外, 对化合物3的吸收光谱进行了无溶剂化(真空)模拟, 发现在考虑溶剂化效应情况下的主要吸收峰与实验值更接近. 化合物4包含一个位于414.7 nm处的高能吸收峰和一个位于552.1 nm处的低能吸收峰, 跃迁形式与化合物3类似, 但是由于其π 链的增长, 导致了体系电荷转移更为明显, 相应的二阶NLO效应也越强.
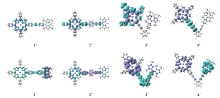
氧化还原对化合物的电子结构必然产生影响, 也势必会影响其NLO性质. 计算了所有化合物的NBO电荷(表4), 并给出自旋密度图(图3). 由表4可知, 化合物1的正电荷主要分布在卟啉和乙炔桥, 负电荷分布在BODIPY, 正负电荷分布相差不大, 而化合物2, 3和4的正电荷分布在卟啉和BODIPY, 负电荷分布在碳硼烷和π 桥上. 氧化态化合物1+~4+的正电荷主要分布在卟啉和BODIPY部分, 说明氧化中心位于卟啉和BODIPY部分, 与图3所示自旋密度一致. 还原态化合物1-和2-的负电荷主要分布在卟啉和BODIPY部分, 说明还原中心位于卟啉和BODIPY; 化合物3-和4-的负电荷主要分布于碳硼烷上, 说明还原中心位于碳硼烷, 自旋密度图表明还原中心也在碳硼烷和π 桥部分.
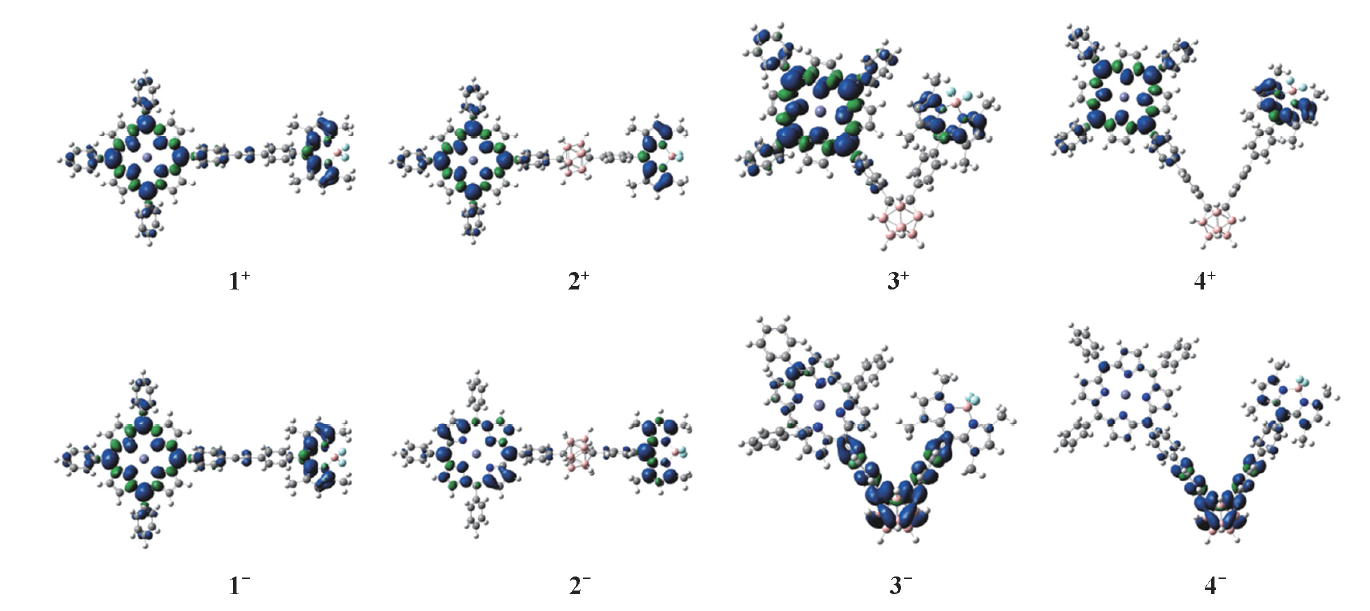 | Fig.3 Spin density plots(Contour value=0.0008) for one-electron oxidized and reduced species of compounds 1— 4 |
| Table 4 NBO charge of all the complexes employing the 6-31G* /LANL2DZ basis set |
采用ω B97XD方法计算氧化态与还原态的β tot值列于表5. 氧化还原态β tot值(除化合物3+)较本征态明显增大, 为了进一步分析氧化还原后化合物β tot值变化的原因, 图4给出化合物氧化还原态的主要激发态的EDDM图. 从图4可知, 氧化或还原后的电荷转移方向相对本征态发生了变化, 氧化态化合物1+和2+发生由π 桥向卟啉以及卟啉内部的电荷转移, 化合物3+电荷转移只发生在卟啉内部, 所以其β tot值较本征态小, 化合物4+表现为卟啉向共轭桥明显的电荷转移; 还原态化合物1-电荷由卟啉、 乙炔向BODIPY转移, 正负电荷间距离较大, 因此其β tot值明显增大, 化合物2-电荷由卟啉向碳硼烷部分转移, 化合物3-表现为明显的碳硼烷向卟啉的电荷转移, 化合物4-电荷转移从BODIPY向卟啉, 相比本征态化合物电荷转移均发生了明显的改变, 且正负电荷距离增大, 这表明氧化还原中电荷转移方式的改变是β tot值变化的主要因素. 因此利用氧化还原可以调控化合物的二阶NLO响应, 为设计优良的NLO转化材料提供了思路.
| Table 5 Computed static first hyperpolarizability for oxidized and reduced of compounds 1— 4 employing the 6-31G* /LANL2DZ basis set |
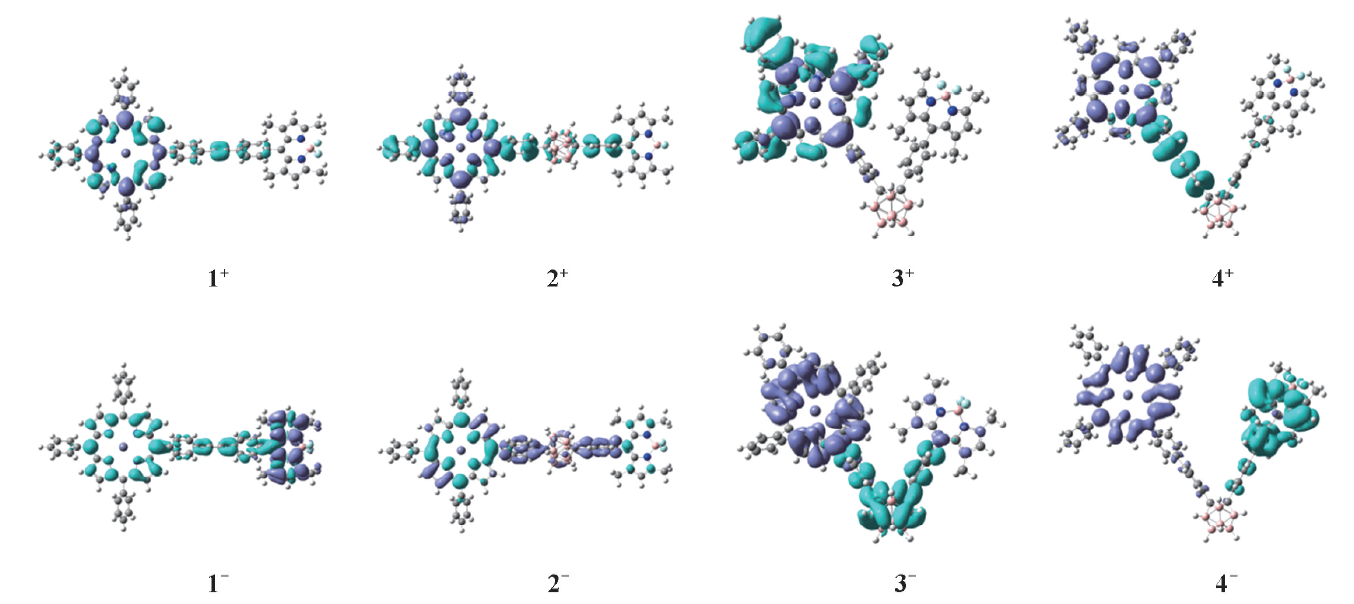 | Fig.4 Electron density difference maps(EDDM) of the main excited state of oxidized and reduced of compounds 1— 4 |
通常, 第一超极化率的实验测量均在激光场条件下进行, 所以在理论计算中考虑频率色散效应非常必要. 为了使计算结果更加接近实验, 因此, 采用CP-DFT ω B97XD方法在6-31G* /LANL2DZ水平上计算了不同频率下化合物1~4的含频第一超极化率. 计算了常见频率ω =0.0239 a.u.(1970 nm)和ω =0.0340 a.u.(1341 nm)的含频第一超极化率β (-ω ; ω , 0)和β (-2ω ; ω , ω )值, 列于表6.
| Table 6 Estimated values of β (-ω ; ω , 0) and β (-2ω ; ω , ω ) at specific frequencies calculated at 6-31G* /LANL2DZ level |
从表6可见, 随着外场频率的增加第一超极化率有所增大, 表明外场频率对化合物的超极化率有一些影响. 化合物1在ω =0.0239 a.u.和ω =0.0340 a.u.时的β (-ω ; ω , 0)值分别约为静态第一超极化率的1.02倍和1.15倍, 在ω =0.0239 a.u.和ω =0.0340 a.u.时的β (-2ω ; ω , ω )值分别约为静态第一超极化率值的1.28倍和1.95倍, 表明频率对β (-2ω ; ω , ω )值的影响比β (-ω ; ω , 0)值较明显一些, 即β (-2ω ; ω , ω )对于化合物表现出较强的色散效应. 化合物3和4也有相同的变化趋势. 化合物2对频率表现出的色散效应更小, 其β (-ω ; ω , 0)值和β (-2ω ; ω , ω )值变化较小. 可见第一超极化率是体系的固有性质, 外场频率在一定范围内变化对第一超极化率影响不大.
采用DFT方法对卟啉-乙炔-BODIPY、 卟啉-碳硼烷-BODIPY三元化合物及其氧化还原态化合物的几何结构、 NBO电荷、 吸收光谱及第一超极化率进行了研究. 结果表明, 桥连基团(乙炔、 碳硼烷)对三元化合物的几何结构影响较小, 而氧化还原对几何结构有一定影响, 还原过程碳硼烷发生扩张. V型化合物明显的电荷转移导致其 β tot值大于直线型化合物, 此外, 延长共轭链可以增大化合物的β tot值, 是提高NLO响应的有效途径. 由NBO电荷分布和电子自旋密度分析得出的氧化/还原中心一致, 结合TD-DFT计算结果及EDDM图可知, 氧化还原过程化合物的电荷转移方式发生改变, 使氧化还原态化合物的β tot值明显变化. 通过计算含频第一超极化率值发现, 一定范围的外场频率对化合物的色散效应较小. 因此, 延长共轭链及氧化还原可作为调节化合物二阶NLO响应的有效手段, 为实验合成此类NLO材料提供理论依据.
支持信息见http://www.cjcu.jlu.edu.cn/CN/10.7503/cjcu20180851.
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|
| [20] |
|
| [21] |
|
| [22] |
|
| [23] |
|
| [24] |
|
| [25] |
|
| [26] |
|
| [27] |
|
| [28] |
|
| [29] |
|
| [30] |
|
| [31] |
|
| [32] |
|