

{kind=link}
{kind=link}
{kind=link}
可快速计算水团簇三体作用强度的新方法
[李晓蕾, 王长生 ]
]
]
|
|


联系人简介: 王长生, 男, 博士, 教授, 博士生导师, 主要从事理论与计算化学研究. E-mail: chwangcs@lnnu.edu.cn
将水分子视为由2个O—H键偶极构成, 再将水分子间的三体作用视为长程诱导作用和短程校正之和, 使用Thole模型计算长程诱导作用, 通过同时考虑不同水分子间的置换和同一个水分子中2个键偶极间的置换计算短程校正, 从而提出了一个可快速计算水团簇三体作用强度的新方法. 根据已报道的12347个水三聚体的结构和CCSD(T)三体作用能, 确定了该方法所需参数. 将该方法和所确定的参数应用于67个水团簇体系, 计算这些体系的三体作用能, 并与CCSD(T), MP2, M06-2X方法的计算结果进行比较. 结果表明, 相对于CCSD(T)方法的总三体作用能, 本文方法的均方根偏差(RMSD)仅为3.32 kJ/mol, 平均相对偏差(MRD)仅为2.43%; 对较大水团簇体系, 该方法计算精度稍优于MP2方法, 明显优于M06-2X方法, 并且更快捷高效.
A method was proposed to calculate the three-body interaction energies in water clusters. The O—H bond of a water molecule was regarded as a bond-dipole, and the three-body interaction energy were expressed as the sum of long-range induction and short-range correction. The long-range induction was treated by using Thole model, and the short-range correction was derived by considering both permutations among different water molecules and permutations between the two dipoles within each molecule. The parameters needed in the method were determined by fitting to the CCSD(T)/aug-cc-pVTZ three-body interaction energies of 12347 water trimers taken from the reference. The accuracy of the method is validated through comparing the three-body interaction energies of 67 water clusters ranging from trimer to 30-mer obtained by CCSD(T) method, MP2 method, and M06-2X method. The method can reproduce the CCSD(T)/aug-cc-pVDZ total three-body interaction energies, with the RMSD of 3.32 kJ/mol and the MRD of 2.43%, slightly better than the MP2 method, and outperforms the M06-2X method for large water clusters. The method is helpful in simulating the dynamic properties of water and other biomolecules.
蛋白质核酸等生物分子体系中以及较大水团簇体系中存在着大量的水分子, 这些水分子间存在着大量的多体作用. 研究发现, 二体作用对水团簇稳定性的贡献最大, 三体作用对水团簇稳定性的贡献也达20%以上[1~4], 因此仅仅准确描述分子间的二体作用不足以保证对生物分子体系稳定性的准确预测, 准确计算水分子间存在的诸多三体作用强度十分重要. Paesani等[5]发现, 对于水团簇体系, 使用CCSD(T)/aug-cc-pVTZ方法结合三体基函数(Trimer basis)计算得到的三体作用能与CCSD(T)/CBS的计算结果相近. Bettens等[6]发现, 使用MP2/aug-cc-pVDZ方法结合三体基函数可以得到与MP2/CBS方法精度相当的三体作用能. 但是当水团簇较大时, 团簇所含的三体作用数目急剧增加(如1个由40个水分子构成的水团簇含有40× 39× 38/6=9880个三体作用), 使用高精度从头算量子化学方法计算水团簇的三体作用能会变得十分昂贵. 因此建立一种可快速准确预测水团簇三体作用能的方法十分必要.
本文提出了一种可快速准确计算水团簇三体作用强度的新方法. 将水团簇中的每个三体作用能表述为长程诱导作用和短程校正之和, 长程诱导作用由Thole模型计算获得, 短程校正项由关于键偶极和水分子的置换不变量构建. 计算结果表明, 该方法不但具有较高的计算精度, 并且非常高效快捷, 有望在生物分子体系的动力学模拟中发挥作用.
将水分子中的每个O— H键视作一个键偶极, 并将水团簇中的每个三体作用能V3B(kJ/mol)视为长程诱导作用
仿照多体展开理论对三体作用能的定义[6, 7], 由水团簇中第m, n, l个水分子组成的三聚体的长程诱导作用
式中:
式中: Ei(N/C)为位点i处的电场; μ i(C· m)为诱导点偶极; α (C· m2· V-1)为原子极化率; T(C-2· J· m-2)为相互作用矩阵; M(C· m)为固有多极矩矢量.
Bowman等[13]利用关于原子的和分子的置换不变量构建了多维势函数, 预测甲烷-水-水复合物的三体作用能. 受此启发, 将水分子看成2个O— H键偶极, 构建关于化学键的置换不变量, 进而计算短程校正
式中: s为开关函数; κ p为关于键偶极和水分子的置换不变量; υ p为参数.
1.2.1 置换不变量κ p的构建 将置换不变量κ p表述为与属于不同分子的2个键偶极间距r有关的e指数函数, 将e指数函数的指数部分包含n个r的置换不变量称为n阶置换不变量. 对一个水的三聚体, 若考虑到六阶置换, 可得2个二阶置换不变量(κ 1~κ 2)、 10个三阶置换不变量(κ 3~κ 12)、 37个四阶置换不变量(κ 13~κ 49)、 106个五阶置换不变量(κ 50~κ 155)及298个六阶置换不变量(κ 156~κ 453), 共计453个置换不变量.
以水团簇中第m, n, l个水分子组成的三聚体为例, 设m1和m2为第m个水分子的2个O— H键偶极, n1和n2为第n个水分子的2个O— H键偶极, l1和l2为第l个水分子的2个O— H键偶极. 仅考虑分子内键偶极间的置换以及3个水分子间的置换, 可得如下所示的一个二阶置换不变量κ 1:
式中: k2和
式中:
式(6)中每项均涉及a, b, c 3个键偶极, 它们分属于3个不同的水分子. 若将式(6)中括号内第一项作为水分子置换的基准, 置换水分子后得第二、 三项, 在此基础上对分子内键偶极的置换求和, 可得κ 1. 若定义Mp为水分子的置换算符, 定义Dp为分子内键偶极的置换算符, 则式(6)可表述为
同理, 三阶置换不变量κ 3、 四阶置换不变量κ 13、 五阶置换不变量κ 50和六阶置换不变量κ 156可分别表述为
式中, kn(n=2~6)和
1.2.2 开关函数s 三体作用强度V3B随三聚体中任意一个水分子的远离而趋近于0. 在保证计算精度的前提下, 使用合适的开关函数, 可去除与分子间距离较远的体系的计算, 减少计算量. 受Paesani等[5]和Li等[23]工作的启发, 将开关函数s设为,
式中: Rij为i, j 2个水分子的O原子间距; Rcut和Δ R为参数; xij为2个水分子的权函数(Weight function). 开关函数s随三聚体中任意一个水分子的远离而逐渐变小, 当有2个以上Rij≥ Rcut+Δ R时, s=0, 则短程作用
对于任意水三聚体结构, 使用所提出的方法计算三体作用能V3B, 需已知开关函数s中的参数Rcut和Δ R, 短程校正项中的453个组合系数υ p(p=1~453), 参数kn(n=2~6)和
选取文献[5]报道的12347个水三聚体结构和CCSD(T)/aug-cc-pVTZ三体作用能作为标准数据, 改变非线性参数Rcut, Δ R, kn(n=2~6)和
由N个水分子构成的团簇中共包含
为了检测所提出方法的准确性, 使用该方法和所确定的参数计算了67个不同水团簇的总三体作用能以及三体作用能随距离和角度变化的能量曲线, 并与使用CCSD(T)/aug-cc-pVDZ方法结合三体基函数的结果进行了对比. 由表S1(见本文支持信息)可见, 使用CCSD(T)/aug-cc-pVDZ方法结合三体基函数、 CCSD(T)/aug-cc-pVTZ方法结合三体基函数计算的总三体作用能差别很小. 为减少计算量, 选择CCSD(T)/aug-cc-pVDZ方法结合三体基函数[简称CCSD(T)方法]的三体作用能作为标准数据. 为了比较, 还给出了使用MP2/aug-cc-pVDZ方法结合三体基函数(简称MP2方法)以及M06-2X/jul-cc-pVTZ方法(jul-cc-pVTZ表示O原子使用aug-cc-pVTZ基组、 H原子使用cc-pVTZ基组[24])结合三体基函数(简称M06-2X方法)计算得到的总三体作用能. 所有从头算均使用Gaussian 09程序包[25]完成.
表1表4给出了不同方法计算得到的67个水团簇的总三体作用能. 其中Δ 为各方法计算的总三体作用能与CCSD(T)结果间的绝对误差, δ 为各方法计算的总三体作用能与CCSD(T)结果间的相对误差.
| Table 1 Calculated total three-body interaction energies(E3B) of 21 small water clustersa |
2.1.1 较小水团簇 表1给出不同方法计算得到的(H2O)n(n=3~6)的总三体作用能. 其中, (H2O)3的结构和CCSD(T)/CBS三体作用能取自文献[26], (H2O)n(n=4~6)的结构和CCSD(T)/aug-cc-pVTZ总三体作用能取自文献[27].
由表1可知, 对这21个较小水团簇, 本文方法计算的总三体作用能与CCSD(T)结果间的最大绝对误差(MAD)在计算六聚体水团簇6w-boat2时产生, 绝对误差为1.96 kJ/mol, 相对误差为4.13%. 对这21个水团簇, 与CCSD(T)方法的结果对比, 本文方法的RMSD为1.02 kJ/mol, 平均相对误差(MRD)为2.86%; MP2方法的RMSD为0.74 kJ/mol, MRD为2.12%; M06-2X方法的RMSD为1.68 kJ/mol, MRD为6.11%; 以上结果表明, 对于这21个较小水团簇, 本文方法与MP2方法计算精度相当, 优于M06-2X方法.
2.1.2 中等水团簇 表2给出了不同方法计算的23个中等尺寸水团簇的总三体作用能. 其中, (H2O)10的结构取自文献[28], 其余水团簇的结构均取自文献[29].
| Table 2 Calculated total three-body interaction energies of 23 medium-sized water clustersa |
由表2可见, 对于这23个水团簇, 本文方法计算的总三体作用能与CCSD(T)结果间的最大绝对误差在计算水二十一聚体第2个构型(21w-2)时产生, 绝对误差为5.65 kJ/mol, 相对误差为3.06%. 由表2还可知, 与CCSD(T)方法的总三体作用能相比, 按3种方法的RMSD, MAD, MRD大小顺序均为本文方法< MP2方法< M06-2X方法. 可见, 本文方法对中等大小水团簇三体作用能的计算精度稍优于MP2方法, 明显优于M06-2X方法.
2.1.3 纳米尺寸水团簇 Truhlar等[24]将至少有一对核间距大于1.0 nm的水团簇定义为水纳米粒子(Water nanoparticle). 相比小的水团簇, 水纳米粒子的成键性质与大量水体系更为接近[24]. 为了考察模型对大量水体系的适用性, 计算了23个水纳米粒子的总三体作用能(见表3). (H2O)16的结构取自文献[30], (H2O)20和(H2O)30的结构取自文献[28], 其余水团簇的结构均取自文献[29]. 对23个水纳米粒子, 与CCSD(T)结果相比, 本文方法的最大绝对误差在计算水二十二聚体[(H2O)22, 22w-1]时产生, 为10.17 kJ/mol, 但相对误差为6.17%.
| Table 3 Calculated total three-body interaction energies of 23 water nanoparticlesa |
与CCSD(T)结果相比, 3种方法的RMSD, MAD, MRD大小顺序均为本文方法< MP2方法< M06-2X方法. 可见, 本文方法对纳米尺寸水团簇三体作用能的计算精度稍优于MP2方法, 明显优于M06-2X方法.
上述67个水团簇中, (H2O)n(n=11~15, 17~19, 21~22)的结构取自文献[29], 该文献报道了WHBB方法的总三体作用能. WHBB方法使用H原子和O原子构建二至六阶置换不变量, 共涉及5894项. 本文方法使用O— H键偶极构建二至六阶置换不变量, 仅涉及453项. 因此本文方法大大减少了置换不变量的个数. 表4表明, 与CCSD(T)方法的总三体作用能相比, 本文方法的计算精度明显优于WHBB方法.
| Table 4 Calculated total three-body interaction energies of 18 water clusters |
表5给出了不同方法相对于CCSD(T)总三体作用能的误差统计. 图1为本文方法的总三体作用能与CCSD(T)结果间的相关性图.
| Table 5 Deviations of different methods with respect to the CCSD(T) three-body interaction energies of 67 water clusters |
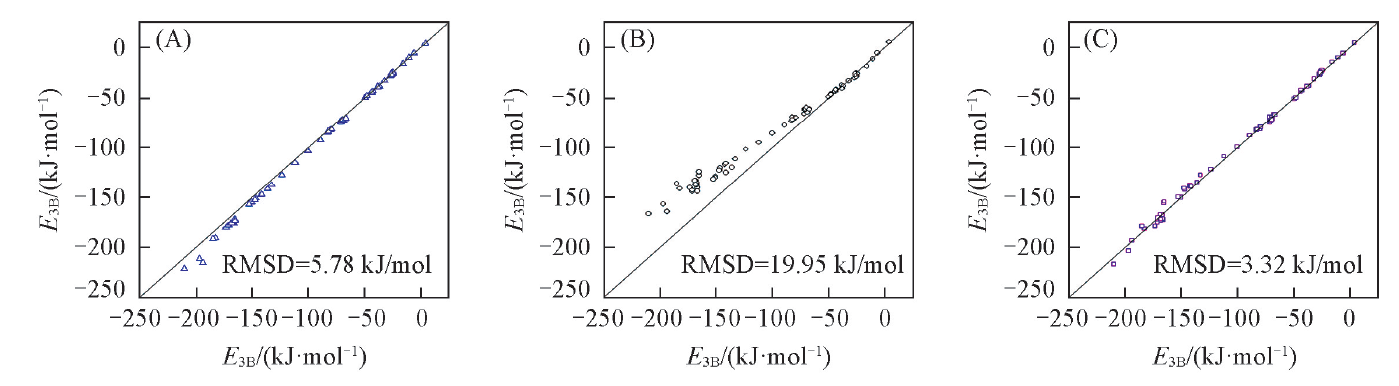 | Fig.1 Correlation plots for the total three-body interaction energies of the 67 water clusters (A) MP2/aug-cc-pVDZ; (B) M06-2X/jul-cc-pVTZ; (C) this work. |
表5和图1进一步表明本文方法对水团簇三体作用能的计算精度稍优于MP2方法, 明显优于M06-2X方法.
图2为水三聚体三体作用能随2个O原子间的距离
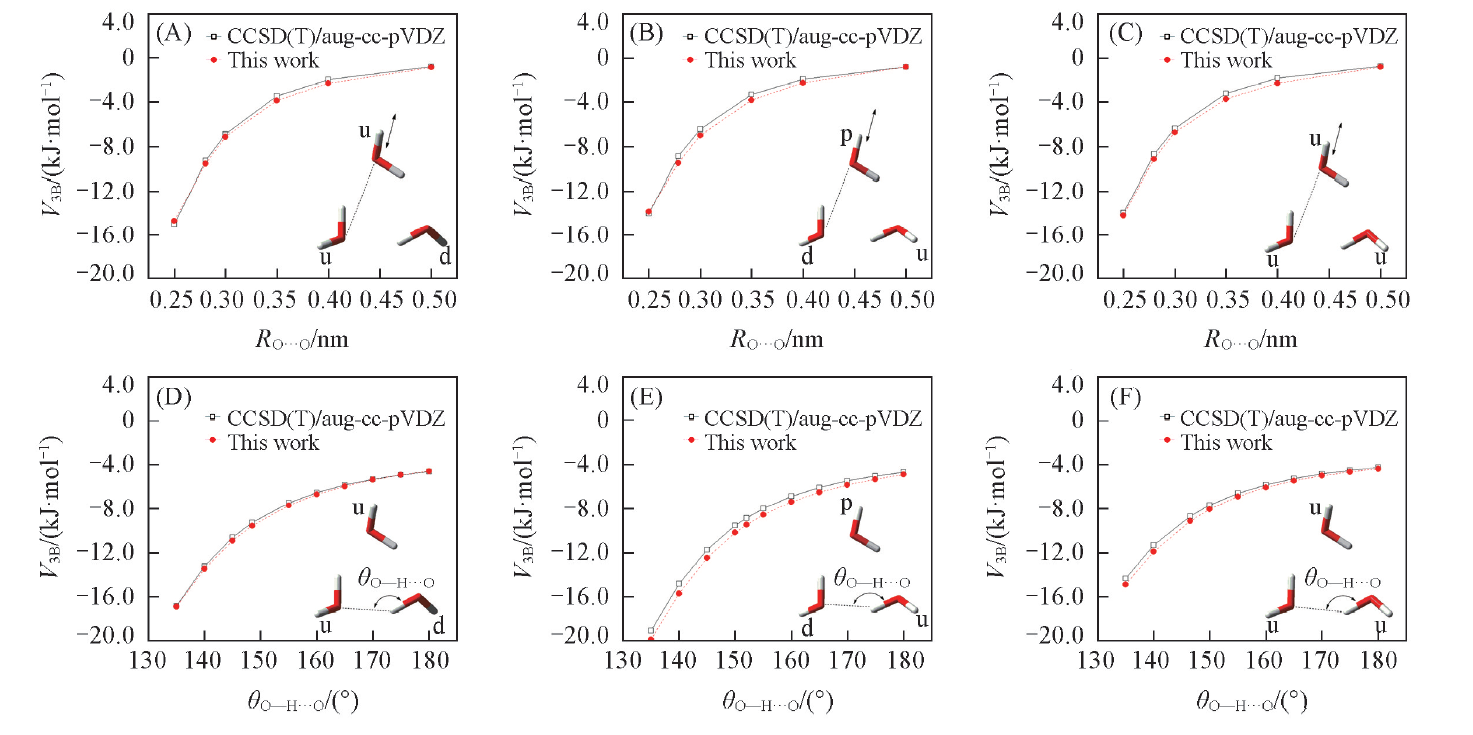 | Fig.2 Three-body interaction energy curves with respect to the intermolecular distances(A— C) and angles(D— F) |
保持2个三元水环的构型不变, 改变2个环中心间的距离
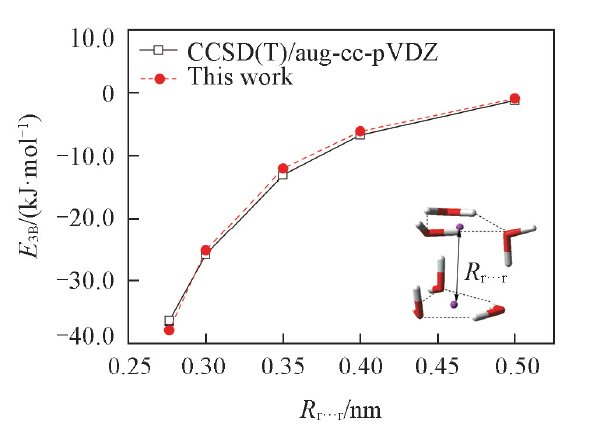 | Fig.3 Total three-body interaction energy curves of the prism hexamer |
由图2和图3可见, 本文方法可以较好地描述三体作用强度随结构的变化.
比较了不同方法在配置为16核芯CPU(Intel Xeon E5-2670)、 96 GB内存和4 TB硬盘服务器上的计算效率. 对于由5个水分子构成的水团簇5w-1的总三体作用能计算结果表明, 使用CCSD(T)方法所需CPU时间为16769 s, 使用MP2方法所需CPU时间为1571 s, 使用M06-2X方法所需CPU时间为11133 s, 使用本文方法所需CPU时间仅为3 s. 对于由10个水分子构成的水团簇10w-1的总三体作用能计算结果表明, 使用CCSD(T)方法、 MP2方法、 M06-2X方法及本文方法计算该团簇总三体作用能所需CPU时间依次为200627, 18476, 137629和3 s. 可见, 本文方法的计算效率远高于CCSD(T), MP2和M06-2X方法, 并且体系越大, 本文方法的计算优势越明显.
提出了一个可快速准确计算水团簇三体作用能的新方法. 使用化学键偶极构建置换不变量, 与使用原子构建置换不变量的WHBB方法相比, 该方法大大地减少了置换不变量的个数, 同时提高了计算精度. 计算结果表明, 该方法不仅可以准确地预测CCSD(T)方法的三体作用能, 而且能够很好地描述三体作用能随水团簇构型的变化. 对较大水团簇三体作用能的计算结果表明, 该方法的计算精度稍优于MP2方法, 明显优于M06-2X方法, 并且运算速度具有明显优势, 体系越大优势越明显.
支持信息见http://www.cjcu.jlu.edu.cn/CN/10.7503/cjcu20190038.
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|
| [20] |
|
| [21] |
|
| [22] |
|
| [23] |
|
| [24] |
|
| [25] |
|
| [26] |
|
| [27] |
|
| [28] |
|
| [29] |
|
| [30] |
|