{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
氧化镧表面反应性对甲烷和氧分子活化的影响
[程文敏, 夏文生 , 万惠霖]
, 万惠霖]
, 万惠霖]
|
|


联系人简介: 夏文生, 男, 博士, 教授, 主要从事催化和理论化学研究. E-mail: wsxia@xmu.edu.cn
以氧化镧催化剂在甲烷氧化偶联(OCM)反应中的结构敏感性实验研究为基础, 采用周期性密度泛函理论(DFT)计算研究氧化镧(001), (110)和(100)3个晶面及OCM反应物分子甲烷和氧在其上的吸附、 活化和解离. 结果表明, 氧化镧(001), (110)和(100)3个晶面的表面能大小顺序为(110)>(100)>(001), 3个晶面的价带和导带间隙大小顺序为(110)<(100)<(001), 即(001)是3个晶面中最稳定的晶面, 而(110)则是最活泼的晶面. 甲烷分子在氧化镧(001), (110)和(100)晶面上的吸附很弱(0.03 eV), H—CH3解离吸附能分别为2.16, 0.68和0.90 eV, 解离反应的难易性与晶面的活性顺序一致; 而氧分子在氧化镧(001), (110)和(100)晶面上的分子吸附能分别为-0.04, -0.31和-0.12 eV, 解离吸附能分别为1.22, 0.53和1.52 eV, 即氧化镧晶面结构对氧分子吸附具有明显的影响, 其中, (001)晶面上吸附最弱, (110)晶面上吸附最强, 以致O—O在(110)晶面上可以较低能垒(0.53 eV)解离, 形成亲电的过氧物种. 由于氧分子在氧化镧表面的吸附较甲烷分子强, 因此, 氧化镧在OCM反应中结构敏感性应与氧分子的吸附和活化密切相关. 甲烷和氧分子在氧化镧表面上活化的本质源自于电子自表面流向甲烷和氧分子的反键轨道, 且表面结构的改变会导致不同强度的电子流动驱动.
The properties of lanthanum oxide facets[(001),(110) and (100)] and the adsorption, activation and dissociation of methane and oxygen on them were studied by means of periodical density functional theory(DFT), based on the previous experimental investigation upon structure sensitivity of lanthanum oxide in Oxidative coupling of methane(OCM). The result shows that surface energy of lanthanum oxide facets decreases in the order of (110)>(100)>(001), and the energy gap between valence and conduction bands follows the order of (110)<(100)<(001), meaning that the facet (001) is most stable and (110) is most reactive among the three facets. The associative adsorption of methane on lanthanum oxide is very weak( ca. 0.03 eV), and the barriers for H—CH3 on lanthanum oxide (001), (110) and (100) surfaces are 2.16, 0.68 and 0.90 eV, respectively, showing the reactivity of the facets is matched with the dissociation of methane. On the other hand, the associative adsorption energy of oxygen on lanthanum oxide (001), (110) and (100) surfaces is -0.04, -0.31 and -0.12 eV, respectively, and the dissociation barrier is 1.22, 0.53 and 1.52 eV, respectively. The oxygen adsorption is greatly influenced by the varied structures of lanthanum oxide. The interaction of oxygen with the facet (001) is the weakest, but the strongest with the facet (110) among the three facets of lanthanum oxide, which promotes O—O bond cleavage at lower barrier(0.53 eV) on the facet (110) to lead to the formation of electrophilic peroxide species. As oxygen adsorption on lanthanum oxide is stronger than methane, the structure sensitivity of lanthanum oxide in OCM should be closely associated with oxygen adsorption and activation. In addition, the mechanism of methane and oxygen activation on lanthanum oxide can be ascribed to the transfer of electrons from the surface to their antibonding orbitals, which implies that varied structures of the lanthanum oxide surfaces can change the driving forces on the electron transfer.
随着能源探测和开采技术的发展, 天然气、 页岩气和可燃冰(甲烷为主要成分, 具有较高的燃烧热值和氢碳比)将成为替代逐渐枯竭的煤和石油资源, 缓解能源和环境紧张态势的重要能源. 在此背景下, 甲烷的转化和利用研究受到了高度瞩目[1~3]. 甲烷催化转化为含氧有机物[4, 5, 6]、 C2+烃类[7, 8, 9]等富有济价值的化学品, 被认为是甲烷转化和利用的有效途径.
由于甲烷分子的化学惰性, 其活化常需在较苛刻(如高温)条件下进行, 而目标产物分子均较甲烷活泼, 易发生进一步的深度转化, 因此甲烷定向转化实际上是一个极富挑战性的动力学控制过程. 鉴于此, 研究者试图寻找和设计能在较低温度下活化甲烷分子的催化剂以提高甲烷转化反应的效率, 如纳米催化剂的设计及其在该领域中的应用. Senkan等[10]将La2O3-CeO2纳米纤维与La2O3-CeO2粉末状催化剂进行对比发现, 前者在更低的温度(470 ℃)下即可进行甲烷氧化偶联反应. Rodriguez等[11]制备了CeOx/Cu2O催化剂, 发现其在有水存在的情况下, 可将甲烷在低温(约180 ℃)下高选择性地转化为甲醇. Bao等[12]设计和制备了石墨烯限域3d过渡金属(锰、 铁、 钴、 镍、 铜)催化材料, 发现石墨烯限域的单铁中心在室温下, 以双氧水为氧化剂, 可直接将甲烷催化转化为高附加值的C1含氧化合物. 上述研究结果表明, 甲烷C— H的活化也可能在较温和的条件下进行而改善产物的选择性.
本课题组[13]曾对甲烷氧化偶联(OCM)反应的纳米催化剂的形貌和结构敏感效应进行了研究. 结果表明, 棒状-H碳酸氧镧、 氧化镧, 与片状-P和另一棒状-Href相比, 能呈现更高的OCM催化性能(甲烷转化率和C2选择性). 棒状-H在温度低至420450 ℃时即呈较良好的OCM催化性能, 片状-P上OCM反应温度需在500 ℃以上, 而棒状-Href上则几乎没有OCM催化性能. 同时发现, 这些样品的性能差异源于其优先暴露晶面[(110), (100)和(001)等]的显著不同, 晶面活性的顺序为(110)> (100)> (001).
为了深入认识这一行为的本质, 本文从密度泛函理论计算角度出发研究OCM反应底物分子(甲烷和氧)在氧化镧晶面上的吸附、 活化等行为, 以期对OCM反应催化剂的设计提供新的认识.
1.1.1 体相结构模型 氧化镧晶体属六方晶系, 空间群为P
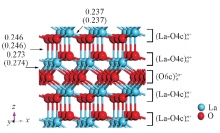
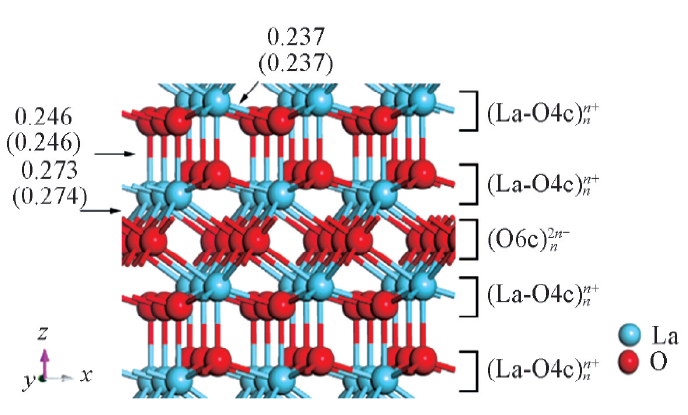 | Fig.1 Structures for bulk La2O3 and the experimental and calculated(in parentheses) values for the three unique La-O bond lengths in nm O4c and O6 crepresent 4- and 6-coordinated oxygen atoms, respectively. |
La2O3晶胞参数的实验数据为a=b=0.394 nm, c=0.614 nm[14]; 而计算所得晶胞参数为a=b=0.395 nm, c=0.616 nm, α =β =90.0° , γ =120.0° , 即实验与计算结果相符.
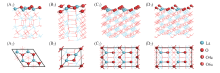
1.1.2 表面结构模型 采用平板模型对La2O3(001), (110)和(100)晶面进行模拟, 如图2所示. 其中, (001)晶面: 选用2× 2超胞模型, 超胞厚度为10个原子层, 优化后的结构显示氧优先暴露于表面; (110)晶面: 选用1× 1超胞模型, 厚度为7个原子层; (100)晶面: 选用2× 2超胞模型, 厚度为12个原子层, 鉴于此表面为极性面的缘故, 选择了将第一层表面一半的氧原子移到底层以消除偶极矩存在带来的影响[15], 图2(C1, C2)和(D1, D2)为矫正前后的(100)晶面模型.
为避免周期性平板间的相互作用, 在超胞间加入了1.5 nm的真空层.
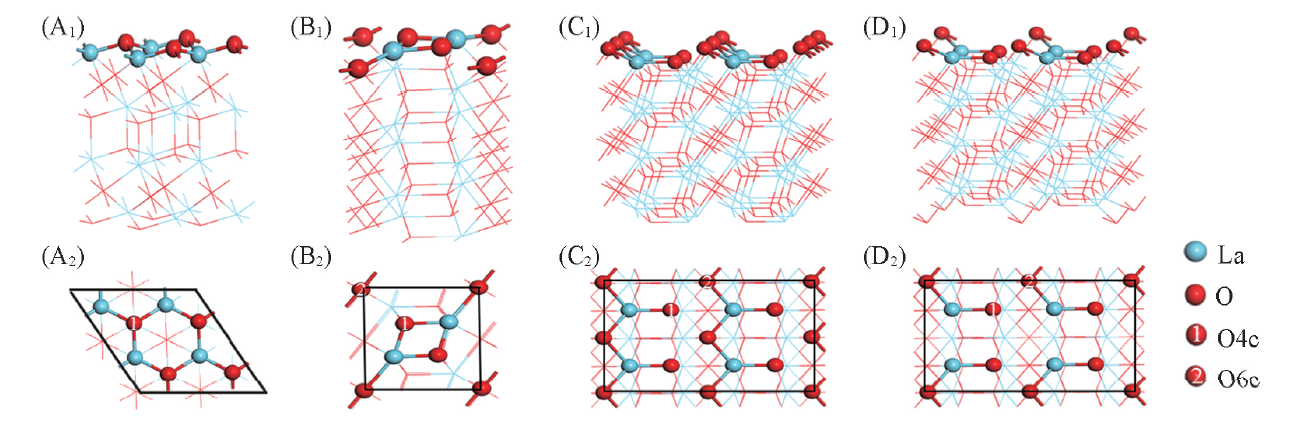 | Fig.2 Side(A1— D1) and top(A2— D2) views of La2O3 surfaces (A1, A2)(2× 2)(001); (B1, B2)(1× 1)(110); (C1, C2)(2× 2)(100), before dipole correction; (D1, D2)(2× 2)(100), after dipole correction; O4c and O6c represent 4- and 6-coordinated oxygen atoms, respectively. |
采用基于自旋极化的密度泛函理论方法, 通过VASP(Vienna ab initio simulation package)软件包[16]完成计算. 离子和价电子间的相互作用采用投影缀加波(PAW)法描述[17]. 由于Metiu等[18]的研究已表明未加库仑排斥势(U)时的广义梯度近似(GGA)处理La2O3体系与加U时(GGA+U)[19]的计算结果一致, 故采用GGA的PBE泛函[20]来处理电子间的交换关联势. 经过严格的测试, 平面波截断动能设为500 eV, 原子弛豫力和自洽能量的收敛阈值分别设为0.2 eV/nm和10-5 eV. K空间取点采用MP方案, 网格对(001), (110)晶面取为5× 5× 1, (100)晶面取为3× 3× 1.
分子在表面上的解离过渡态搜寻, 通过采用爬坡弹性带(CI-NEB)法来进行, 其相应的原子弛豫力和自洽能量收敛阈值分别设为0.5 eV/nm和10-5 eV.
计算中, 氧分子吸附在表面上时体系呈三重态, 而解离吸附因过氧化物种的形成, 体系呈单重态, 即计算过程中体系的自旋是非限制的.
表面能的计算如下:
AB分子在表面上非解离吸附能计算如下:
AB分子在表面上解离吸附能计算如下:
式中: γ (eV)为表面能; Eslab(eV)为平板的能量; Ebulk(eV)为固体体相单位晶胞的能量; S(nm2)为平板表面积; n为平板中所含晶胞数; Ead(AB)(eV)和Ead(A-B)(eV)分别为AB分子在表面上的分子吸附能和解离吸附能; E(slab+AB)(eV)和E(slab+A-B))(eV)分别为AB分子的吸附态和解离吸附态的能量; EAB(eV)为AB分子的能量.
体相La2O3中的La为七配位, 标记为La7c, O则分为四配位(占2/3)和六配位(占1/3)2种类型, 分别标记为O4c和O6c; 而对模型表面原子, 为明确起见, 仍以体相配位标记予以区分.
由图2可见, La2O3(001)(2× 2)超胞表面由4个La7c和4个O4c构成, 其配位数分别为6和3; La2O3(110)(1× 1)单胞表面由2个La7c, 2个O4c和1个O6c构成, 其配位数分别为5, 3和4; La2O3(100)(2× 2)超胞表面则由4个La7c, 4个O4c和2个O6c构成, 其配位数分别为4, 3和3. 相对于体相原子, 3个晶面中的(001)晶面原子配位数的下降程度最低, 由此可认为, La2O3(001)晶面相对最稳定. 也可由表面能的计算值高低来予以佐证. 经计算, La2O3(001), (110)和(100)晶面能大小顺序为(001)(4.01 eV/nm2)< (100)(5.78 eV/nm2)< (110)(7.10 eV/nm2). 即La2O3(001)晶面是最稳定的. 与文献报道的实验结果[21]和Illet等[22]的理论计算结果一致.
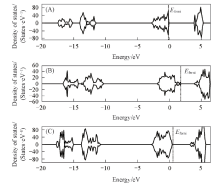
图3给出了La2O3(001), (110)和(100)晶面的态密度, 可见, 其态密度形态相似, (001), (110)和(100)的价带和导带能隙分别约为4.0, 2.1和2.8 eV, 而费米能级则分别约为-0.30, 1.86和0.49 eV, 二者的大小(高低)排序相反. 价、 导带能隙越小(电子云易受外界扰动), 费米能级越高, 表面给电子能力越强, 表面反应性也越强. 因此, 相对而言, (001)晶面给电子能力最弱, 而(110)晶面给电子能力最强. 这3个晶面费米能级大小排序为(001)< (100)< (110), 与其表面能的大小排序一致.
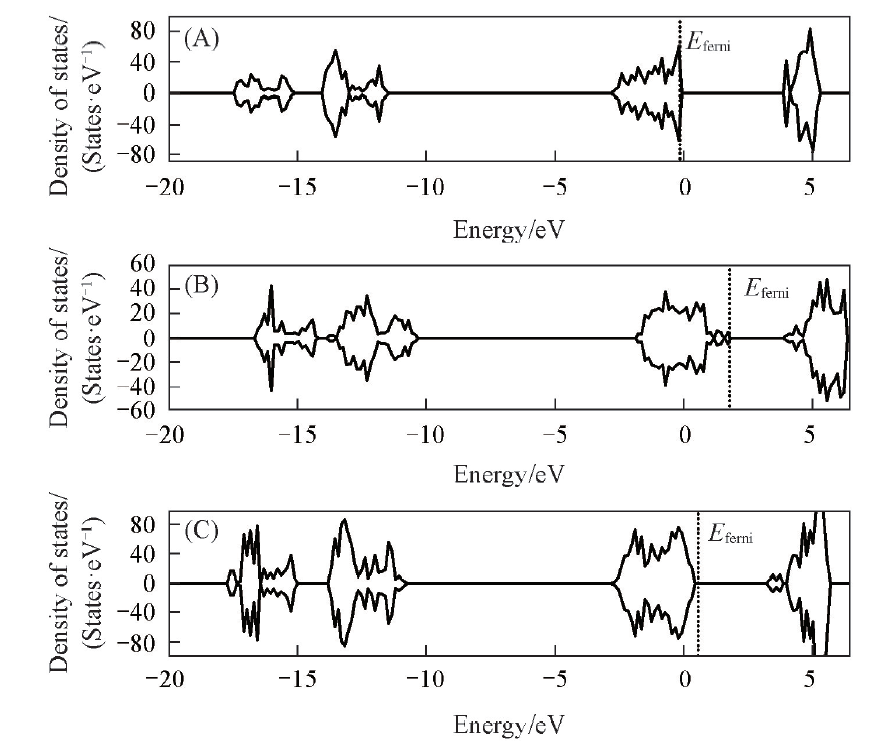 | Fig.3 Density of states(DOS) of La2O3 surfaces (001)(A), (110)(B) and (100)(C) The densities of states above 0 are for orbitals with spin up, and those below 0 are for orbitals with spin down. |
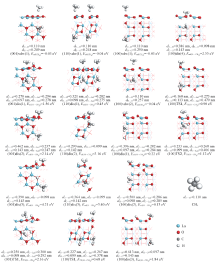
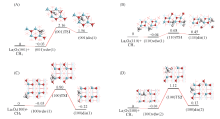
CH4在La2O3(001), (110)和(100)晶面上的分子吸附作用强度微弱. 如图4所示, CH4吸附能约0.03~0.04 eV, 说明CH4与表面几乎没有相互作用, 分子吸附态vdw中H与最邻近表面原子间距为0.249~0.257 nm, 而C— H键长约0.110 nm, 与自由甲烷分子C— H键长值大小相当, 几乎没有差异.
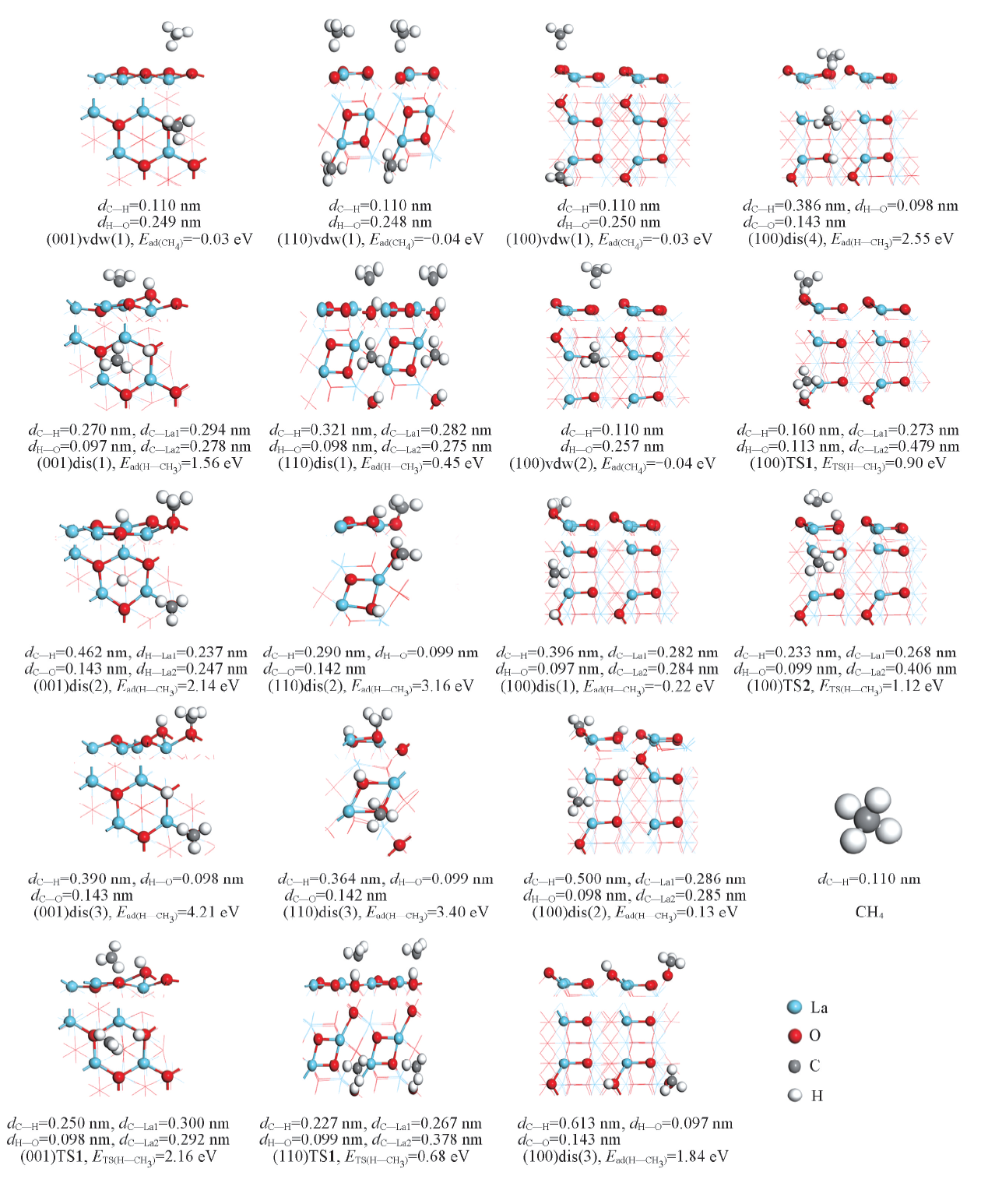 | Fig.4 Structures, energies and related bond length of associative(vdw) and dissociative(dis) adsorption states and corresponding transition states(TS) for CH4 on La2O3 surfaces(001), (110) and (100) |
CH4在La2O3(001), (110)和(100)晶面上的解离吸附, 因这些表面的结构差异和解离碎片CH3, H在表面上的落位点的组合, 必然会出现几种不同结构和作用强度(甲烷解离吸附能)的模式. 由图4可见, (001)晶面上存在(001)dis(1), (001)dis(2)和(001)dis(3) 3种解离吸附模式, 解离吸附能分别为1.56, 2.14和4.21 eV, 与Metiu等[23]的计算结果(1.62, 2.11和4.18 eV)相符, 其中, 模式(001)dis(1)吸附能最低; (110)晶面上CH4的解离吸附也存在(110)dis(1), (110)dis(2)和(110)dis(3) 3种模式, 相应的解离吸附能分别为0.45, 3.16和3.40 eV, (110)dis(1)模式具有显著的能量优势; (100)晶面上CH4的4种解离吸附模式(100)dis(1), (100)dis(2), (100)dis(3)和(100)dis(4)对应的解离吸附能分别为-0.22, 0.13, 1.84和2.55 eV, 模式(100)dis(1)和(100)dis(2)具有相对较高的能量优势. 值得注意的是, 吸附能较低的模式均为解离碎片CH3落于邻近La-La桥的位点上, 而落于单个O位点上的吸附能值则均较高. 比较3个晶面上CH4解离吸附的情况可发现, CH4在(001)晶面上的解离吸附能最高, 在(100)晶面上的解离吸附能最低, 即无法据此有效解释实验观察到的OCM反应中La2O3的晶面效应.
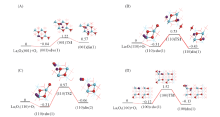
以能量优势的解离吸附模式为基础寻找过渡态, 构建CH4在La2O3表面上发生C— H键裂的势能面示意图, 则可对不同结构表面上甲烷C— H活化的表现给予更深入的认识. 如图5所示, (001)晶面上, 过渡态(001)TS1相对于反应物的能量为2.16 eV, 其结构特征为, 碎片CH3与2个La原子间距分别为0.300和0.292 nm, H与C, O间距分别为0.250和0.098 nm, 而与之关联的解离产物CH3与邻近的2个La原子间的距离分别为0.294和0.278 nm, H与C, O间距分别为0.270和0.097 nm[图4, (001)TS1, (001)dis(1)], 说明解离吸附能高导致了对应的过渡态在能量和几何结构上已很接近于解离产物, 根据Brø nsted-Evans-Polanyi(BEP)关系, 甲烷C— H的活化能垒偏高; (110)晶面上, 过渡态(110)TS1相对于反应物的能量为0.68 eV, 其结构特征为, 碎片CH3与2个La原子间距分别为0.267和0.378 nm, H与C, O间距分别为0.227和0.099 nm, 而与之关联的解离产物CH3与2个La原子间的距离分别为0.275和0.282 nm, H与C, O间距为0.321和0.098 nm(图4), 过渡态结构与解离产物结构相似, 甲烷C— H活化的能垒并不高, 这可能与(110)晶面本身的表面能较高有关; (100)晶面上, 两条能量相当的甲烷C— H活化途径的过渡态(100)TS1[(100)TS2]相对于反应物的能量为0.90(1.12) eV, 其结构特征为, 碎片CH3与2个La原子间距分别为0.273(0.268)和0.479(0.406) nm, H与C, O间距分别为0.160(0.233)和0.113(0.099) nm, 而与之关联的解离产物CH3与2个La原子间的距离分别为0.282(0.285)和0.284(0.286) nm, H与C, O间距为0.396(0.500)和0.097(0.098) nm[图4, (100)TS1, (100)TS2, (100)dis(1), (100)dis(2)], 其中能量较低(0.90 eV)途径的过渡态(100)TS1的H— CH3仍保留较明显的C— H键特征(C— H键长0.160 nm). 因此, 从表面上甲烷C— H活化的能垒高低看, 3个晶面的反应活性高低顺序为(110)> (100)> (001).
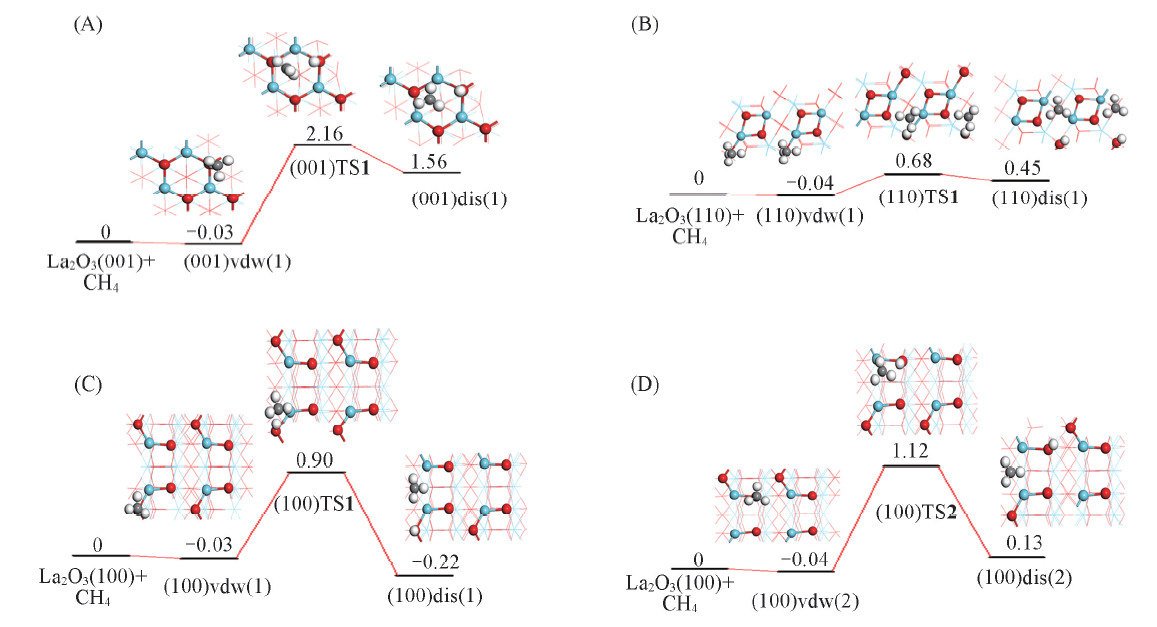 | Fig.5 Diagram of potential energy surface for methane C— H activation on La2O3 surfaces (A) (001), H is bound to O4c; (B) (110), H is bound to O6c; (C) (100), H is bound to O6c; (D) (100), H is bound to O4c. The energy is in eV. |
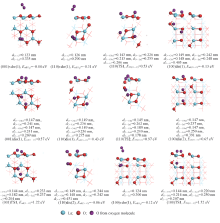
O2与La2O3的作用要大于CH4. 如图6所示, O2在La2O3(001), (110)和(100)晶面上的分子吸附能分别为-0.04, -0.31和-0.12 eV, 呈现出明显的晶面效应, 特别是(110), (100)晶面上的分子吸附能低于CH4的, 且O— O键长(及O与邻近的表面La原子间距)分别为0.123(0.335), 0.126(0.290)和0.124(0.300) nm, 说明O— O键在(001)晶面上所受影响甚微, 但在(110), (100)晶面上则发生一定程度的活化.
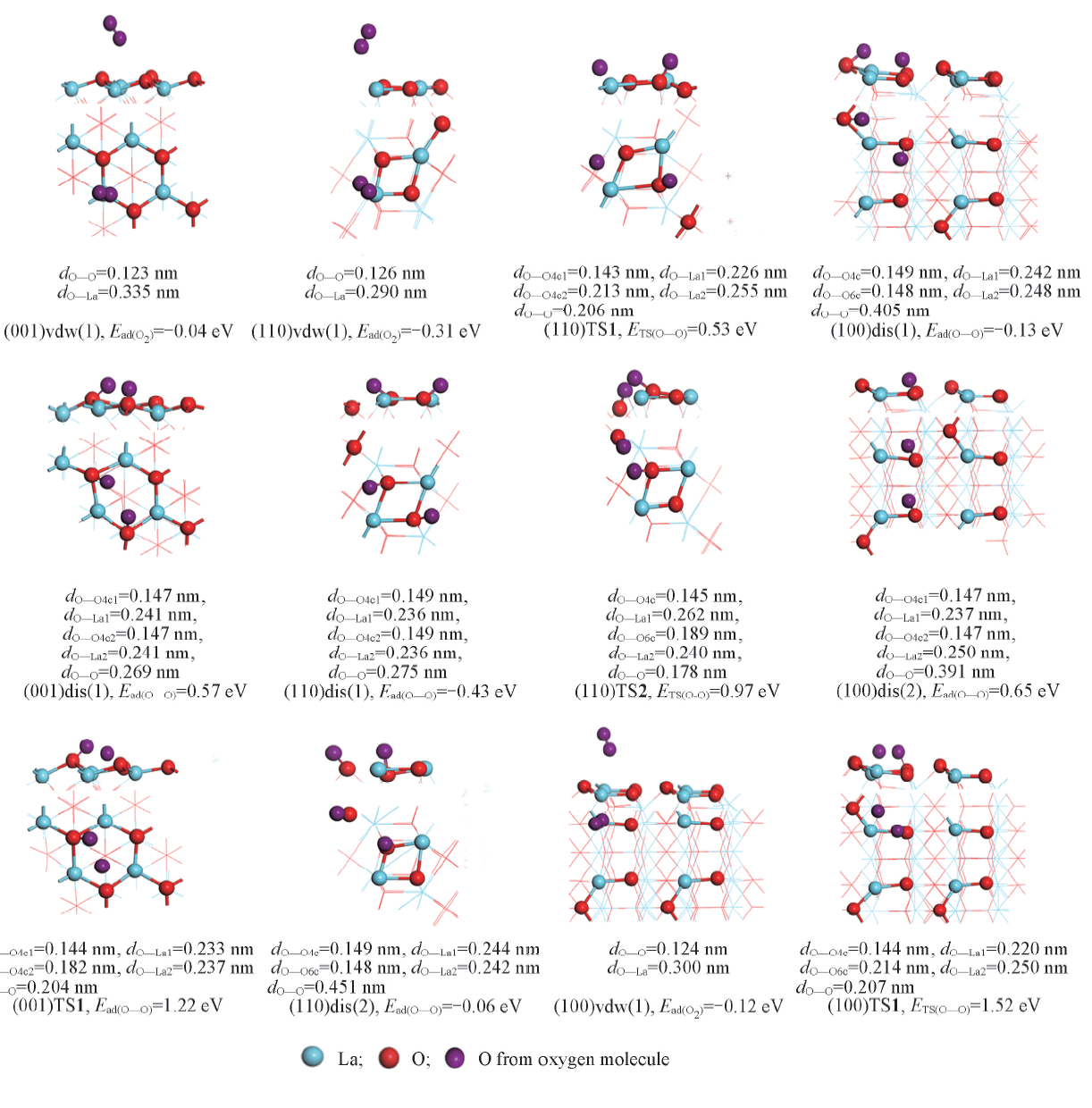 | Fig.6 Structures, energies and related bond length of O2 associative adsorption(vdw), dissociative(dis) adsorption modes and corresponding transition states(TS) on La2O3 surfaces (001), (110) and (100) |
O2在La2O3表面上的解离吸附是通过其与La2O3的表面O成键, 逐步发生O— O键裂, 形成2个(键长约0.147~0.149 nm)新的双氧物种(依照Neurock等[24]指认该物种为过氧物种)而完成的; 由于La2O3表面(001), (110)和(100)的结构差异及表面上O4c和O6c等位点的差异, O2在这些表面上的解离吸附势必会呈现几种结构不同和作用强度(解离吸附能)不同的模式. 如图6所示, (001)晶面上, O2解离吸附仅存在一种模式(001)dis(1), 解离吸附能为0.57 eV; (110)晶面上, O2的解离吸附存在(110)dis(1)和(110)dis(2) 2种模式, 解离吸附能分别为-0.43和-0.06 eV; (100)晶面上, O2的解离吸附也存在 (100)dis(1)和(100)dis(1) 2种模式, 解离吸附能分别为-0.13和0.65 eV. 因此, O2在(001), (110)和(100)晶面上的最优解离吸附能分别为0.57, -0.43和-0.13 eV, 即(110)和(100)晶面上O2解离吸附能为负值, 而(001)晶面上则为正值. 这与本课题组[13]报道的O2-程序升温脱附(O2-TPD)结果相吻合, 优先暴露(001)晶面的样品上未发现氧脱附峰, 而优先暴露(110)晶面的样品上则发现540 ° C处出现归属于亲电氧物种的脱附峰.
图7为La2O3表面上O— O活化的势能面示意图. 可以看出, (001)晶面上, 过渡态(001)TS1相对反应物的能量为1.22 eV, 与Li等[25]的计算结果(1.26 eV)相近, 过渡态(001)TS1的结构特征(图6)为, O— O键断裂的2个O原子与邻近的表面上2个O4c的间距分别为0.144和0.182 nm, 与La原子的间距分别为0.233和0.237 nm, 2个O原子自身的间距则为0.204 nm; 而与之关联的解离产物2个O原子与邻近的表面上2个O4c的间距皆为0.147 nm, 与La的间距皆为0.241 nm, 2个O原子自身的间距则为0.269 nm, 说明较高的解离吸附能值(0.57 eV)导致了过渡态在能量和几何结构上已很接近于解离产物, 根据BEP关系, (001)晶面上O— O能垒偏高. (110)晶面上, 过渡态(110)TS1[(110)TS2]相对反应物的能量为0.53(0.97) eV, 其结构特征(图6)为, O— O键断裂的2个O原子与邻近的表面上2个O4c(O4c, O6c)的间距分别为0.143, 0.213(0.145, 0.189) nm, 与La的间距分别为0.226, 0.255(0.262, 0.240) nm, 2个O原子自身的间距则为0.206(0.178) nm; 而与之关联的解离产物2个O原子与邻近的表面上2个O4c(O4c, O6c)的间距分别为0.149, 0.149(0.148, 0.149) nm, 与La原子的间距分别为0.236, 0.236(0.244, 0.242) nm, 2个O原子自身的间距则为0.275(0.451) nm; 由于解离前后能量差不大(-0.12或0.25 eV), 且解离的O原子与多个La原子接邻(图6), 故(110)晶面上O— O键断裂能相对较低. (100)晶面上, 过渡态(100)TS1相对反应物的能量为1.52 eV, 其结构特征(图6)为, 2个O原子与邻近表面上O4c, O6c的间距分别为0.144和0.214 nm, 与La原子的间距分别为0.220和0.250 nm, 2个O原子自身的间距则为0.207 nm, 而与之关联的解离产物2个O原子与邻近表面上O4c, O6c的间距分别为0.149和0.148 nm, 与La原子的间距分别为0.242和0.248 nm, 2个O原子自身的间距则为0.405 nm; 由于解离前后的能量差不大(-0.01 eV), 过渡态中的键裂O原子与较少的La原子接邻(图6), 而(100)晶面活性不如(110)晶面, 故(100)晶面上O— O键断裂能将不会很低. 因此, La2O3(001), (110)和(100)晶面上O— O活化能大小顺序为(110)(0.53 eV)< (001)(1.22 eV)< (100)(1.52 eV), 即从动力学角度判断, (110)晶面活性最强.
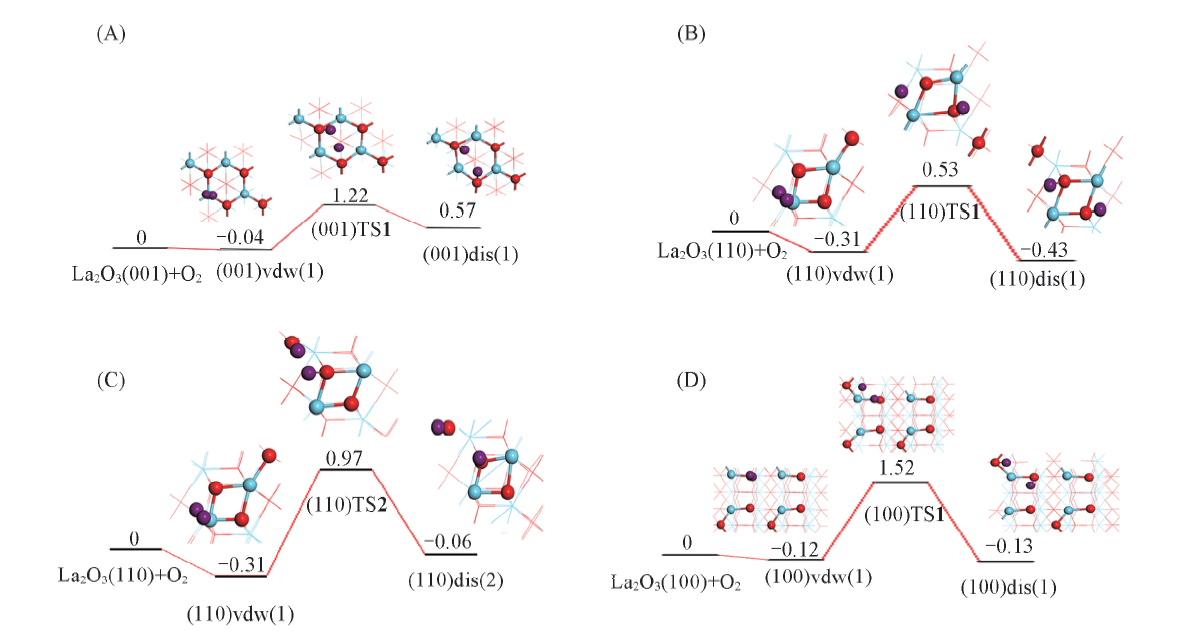 | Fig.7 Diagram of potential energy surface for O— O activation on La2O3 surfaces (A) (001), O is bound to two O4c; (B) (110), O is bound to two O4c; (C) (110), O is bound to O6c/O4c; (D) (100), O is bound to O6c/O4c. The energy is in eV. |
如前所述, CH4在La2O3(001), (110)和(100)晶面上的缔合式吸附强度微弱(微小的吸附能负值), 解离式吸附能仅在(100)晶面上呈现较小的负值, 其它均为正值; 而O2在La2O3表面上的缔合式和解离式吸附则因表面结构呈现不同的情况, 其中, (001)晶面与O2的作用微弱, (110)和(100)晶面与O2的作用较强, 其缔合和解离式吸附能均为负值, 即O— O在(110), (100)晶面上得到了一定程度的活化(表1). 因此, O2在这些表面上的活化程度大于CH4, 且活化难易与表面结构密切相关.
| Table 1 Energy of the preferred associative adsorption states, dissociative adsorption states and transition states relative to reactants(Ead(AB), Ead(A-B), ETS(A-B)) for CH4 and O2 on La2O3 surfaces, and an analysis on Bader charges(qAB, qTS(A-B), qA-B) of the corresponding species |
由表1可知, CH4缔合式吸附在La2O3表面上时, (001), (110)和(100)晶面上分子吸附态的Bader电荷分别为-0.01, -0.02和-0.02 e, 而过渡态(解离吸附态)的Bader电荷则分别为-0.03, -0.10和-0.09 e(-0.10, -0.10和-0.12 e); O2缔合式吸附在La2O3表面上时, (001), (110)和(100)晶面上的分子吸附态的Bader电荷分别为-0.02, -0.24和-0.10 e, 而过渡态(解离吸附态)的Bader电荷则分别为-1.02, -1.05和-1.04 e(-1.19, -1.34和-1.30 e). 由缔合式的分子吸附态到解离过渡态直至解离吸附态, 相关物种的电荷负值呈增大趋势, 说明H— CH3和O— O的活化是因电子由La2O3表面流向CH4和O2的反键轨道所致, 且这种电子流动以促使化学键活化直至解离的驱动力因表面结构而有所不同, 其大小顺序为(110)> (100)> (001), 与前述各表面的表面能高低顺序一致.
由于O— O与La2O3表面的作用无论是从热力学还是动力学角度出发均优于H— CH3, 因此, OCM催化反应应当是通过反应物之一的O2优先吸附于La2O3表面, 形成过氧物种(
已有研究表明, 表面氧物种的存在对甲烷C— H活化具有显著的影响. 如Wang等[26, 27, 28, 29]通过理计算考察了金属表面上的预吸附氧物种(O* ,
采用DFT法计算研究La2O3(001), (110)和(100)晶面的性质及其对甲烷和氧分子的作用发现, La2O3(001), (110)和(100)晶面能的大小排序为(110)> (100)> (001), 与晶面的费米能高低排序一致, 但与其价带-导带间隙大小排序正好相反, 因而导致了这些晶面的不同反应性. 虽然甲烷分子在这些晶面上的吸附很弱, 但其解离吸附能垒高低的顺序与这些晶面的表面能高低呈相反趋势, 即(110)晶面上甲烷C— H键裂的能垒最低, (001)晶面上能垒最高; 然而, 氧分子在这些晶面上的缔合式吸附即具有明显的晶面效应, 吸附能高低顺序与晶面的表面能排序一致, 即氧分子在(001)晶面上的吸附最弱, 而在(110)晶面上的吸附最强, 并在较低能垒(0.53 eV)下促使O— O键裂形成亲电的过氧物种, 从而有助于OCM反应的进行. 因此, OCM反应催化剂La2O3的结构敏感效应与氧分子的吸附和活化密切相关. 甲烷和氧分子在La2O3表面上活化的机制源于电子自表面流向甲烷和氧分子的反键轨道, 而表面结构的差异会导致不同强度的电子流动驱动力.
感谢李圣刚教授(中国科学院低碳转化科学与工程重点实验室)在甲烷解离吸附模式方面的一些建议.
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|
| [20] |
|
| [21] |
|
| [22] |
|
| [23] |
|
| [24] |
|
| [25] |
|
| [26] |
|
| [27] |
|
| [28] |
|
| [29] |
|