
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
MOFs基材料在光催化CO2还原中的应用
[何鹏琛, 周健, 周阿武, 豆义波 , 李建荣]
, 李建荣]
, 李建荣]
|
|


联系人简介: 豆义波, 男, 博士, 副研究员, 主要从事金属有机框架基复合功能材料研究. E-mail: douyb@bjut.edu.cn;李建荣, 男, 博士, 教授, 主要从事新型多孔材料设计制备及应用研究. E-mail: jrli@bjut.edu.cn
系统总结了金属有机框架(MOFs)基材料在光催化还原CO2中的最新研究进展, 其中包括MOFs直接作为光催化剂和作为复合光催化2个主要部分, 讨论了MOFs基光催化剂在催化还原CO2方面展现出的独特优势, 并对MOFs基光催化剂的结构稳定性与CO2转化效率等问题进行讨论与分析, 对未来发展趋势进行了展望.
We summarize the recent progress of metal-organic frameworks(MOFs) used for photocatalytic CO2 reduction, including MOFs employed directly as the photocatalysts for CO2 reduction and as components in hybrid photocatalytic systems for CO2 reduction. Meanwhile, the fascinating features of MOFs-based photocatalysts in CO2 reduction are presented. Finally, the challenges and perspectives regarding of MOFs-based photocatalyst are also discussed.
当前, 全球能源过度消耗和环境日益恶化已成为人类社会面临的严峻问题, 特别是由于二氧化碳(CO2)大量排放引发的一系列环境问题, 如气候恶化、 地球表面温度升高、 两极冰川融化等. 因而, 有效减少大气中CO2的含量, 同时开发环境友好且可再生的新能源成为人们关注的焦点[1~5]. 人们曾尝试将排放的CO2资源化回收转化为具有可利用价值的化学品或燃料(CO, CH4, CH3OH, HCOOH等), 该方法有望在缓解能源危机的同时缓解“ 温室效应” [6]. 目前已经有多种方法用于CO2转化, 如光催化还原[7, 8]、 电化学还原[9, 10]和生物转化[11, 12, 13]等. 在这些方法中, 光催化还原CO2由于具有清洁环保的特点而备受关注. 目前用于还原CO2的光催化剂主要有传统的半导体材料(WO3[14], TiO2[15], , ZnO[16, 17]等)、 贵金属[18](Pt, Pd, Au等)和有机染料光敏剂([Ru(bpy)3]Cl2等[19]). 但是, 传统光催化剂仍受限于光吸收能力较弱、 CO2吸附和活化困难、 电子-空穴易于复合、 能量转化效率低、 稳定性差以及催化剂的带隙能级与太阳光谱之间的差异性等因素的困扰[20, 21, 22], CO2的催化还原效率低以及选择性差等问题亟待解决. 因此, 研究和开发新型高效且稳定的CO2光催化还原剂仍然面临着许多挑战.
金属有机框架(Metal-organic frameworks, MOFs)材料作为一种多孔材料, 通过有机配体与金属离子或金属簇节点之间的配位键连接形成, 在气体储存[23]和分离[24, 25]及催化[26, 27]等领域得到广泛研究. 由于其高孔隙率以及与CO2的相互作用, 在CO2捕获方面也被大量研究[28]. 目前, 部分MOFs在光催化领域表现出良好的催化性能[33, 39, 40]. MOFs作为光催化剂具有如下特点: (1) MOFs自身的高比表面积有助于底物分子在活性位点周围的吸附和富集, 有利于后续过程中的活化和催化转化; (2) MOFs中各个金属-氧单元之间由于有机配体的存在, 形成了独立的类似半导体的结构特征, 有利于电子与空穴的分离[29]; (3) MOFs可以通过筛选不同的有机配体和金属中心, 有效提高太阳光的吸收和利用效率[30]; (4) 可以通过改变晶体结构来促进电荷的分离和转移, 从而抑制电子与空穴的复合; (5) 作为非均相催化剂, MOFs易于从反应体系中分离再循环利用, 有利于延长催化剂的使用寿命并避免产生污染. 本文系统概述了近年来MOFs在光催化还原CO2领域的研究进展, 综述了单一MOFs及后修饰官能团的MOFs材料在光催化CO2方面的研究, 以及MOFs与其它组分形成的复合材料在光催化还原CO2方面的应用, 并对MOFs基材料在光催化CO2领域的发展趋势、 存在的问题及可能解决的方法进行了讨论.
MOFs在CO2还原领域有广阔的应用前景, 其可以设计成具有开放的金属位点、 特定的杂原子和功能化的有机配体的有序结构, 能够有效提高电子-空穴分离效率和载流子的利用效率, 从而提升光催化性能[31]. 多孔性可使MOFs暴露更多的活性位点和通道, 在提高电荷传递效率的同时抑制电子-空穴在体相中的复合, 进而提高其利用效率. 基于以上因素, 人们尝试将不同MOFs用于光催化CO2还原研究. Chen等[32]研究发现, 在可见光下新型Zr-MOF可以有效地将CO2光催化还原成HCOO-, 并且具有较高的转化效率. 该MOF在催化过程中表现出双重途径, 即配体的直接激发和配体到金属簇的电荷转移, 进一步证明了其具有较高的催化性能.
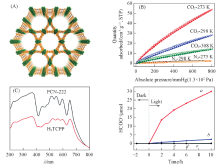
近年来, 卟啉或金属卟啉因具有较高的光捕获效率和催化活性而成为人们关注的焦点. Xu等[33]研究发现, PCN-222[结构见图1(A)]能选择性捕获CO2. 在1× 105 Pa压强下于308, 298和273 K时分别显示出14, 35和58 cm3/g的高CO2吸附量[图1(B)], 这表明PCN-222与CO2之间有较强的相互作用. 此外, PCN-222的紫外-可见光谱(UV-Vis)在200800 nm区域显示出宽的吸收范围[图1(C)]. 研究发现, 随着光照时间的延长, PCN-222表现出显著的光催化活性[图1(D)], 反应10 h后HCOO-的生成量可达30 μ moL, 而且没有检测到其它产物, 表明该催化剂对CO2转化具有高度选择性. 良好的催化活性可能是因为PCN-222是由卟啉配体与Zr簇的有序结合, 可以有效避免电子-空穴复合, 从而提升光催化效率. 同时, PCN-222能够有效捕获CO2, 并且为光还原提供长寿命的电子, 最终表现出的活性远高于H2TCPP配体.
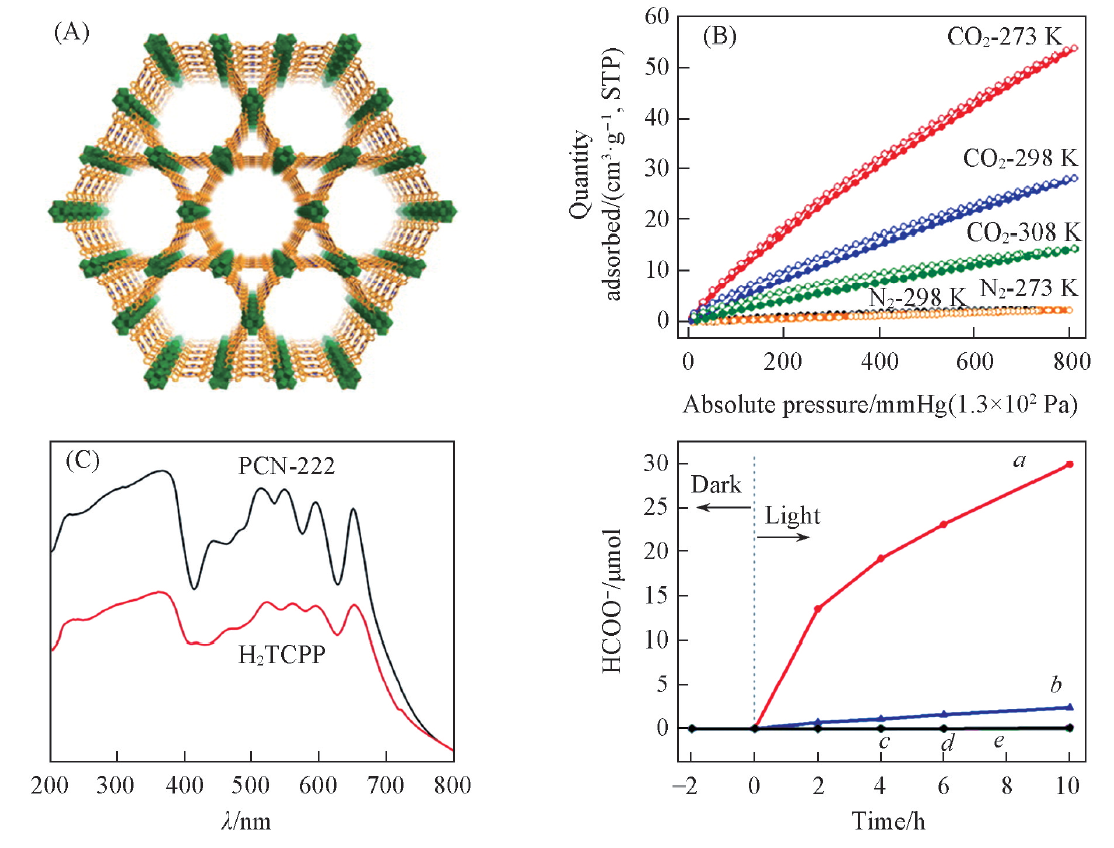 | Fig.1 Structure(A) and CO2 and N2 adsorption isotherms(B) of PCN-222, UV-Vis spectra of PCN-22 and H2TCPP(C) and the amount of HCOO-(D) with PCN-22 as catalyst[33] (D) a. PCN-222; b H2TCPP; c. no PCN-222; d. no TEOA; e. no CO2. Copyright 2015, American Chemical Society. |
Sadeghi等[34]合成了锌基卟啉(Zn/PMOF), 其能够在光照条件下催化还原CO2为CH4. 结果表明, 以Zn/PMOF作光催化剂, 光照4 h后CH4的生成量为10.43 μ mol, 远高于ZnO作光催化剂时生成CH4的量. 同时, Zn/PMOF作光催化剂对CO2还原具有高选择性, 而且具有良好的稳定性和可重复性.
由于铁资源的丰富性和铁基光催化剂的可见光响应性, 以铁为金属中心的MOFs基光催化剂也被广泛研究[35]. Wang等[36]研究了一系列Fe-MOF(MIL-101, MIL-53, MIL-88B)在可见光驱动下催化还原CO2的性能. 结果发现, MIL-101(Fe)的催化效果最佳, 这可能是由于其结构中存在配位不饱和的Fe位点, Fe— O团簇吸收光可以诱导电子从O2-转移到Fe3+形成Fe2+, 进而参与CO2的活化. 此外, 对Fe-MOF进行NH2修饰后光催化活性又明显提高, NH2-MIL-101(Fe)作光催化剂催化还原CO2时生成的HCOO-的量比MIL-101(Fe)约高3倍. 相同条件下, NH2-MIL-53(Fe)和NH2-MIL-88B(Fe)催化还原CO2的能力也明显分别高于MIL-53(Fe)和MIL-88B(Fe). 由于双重催化途径的协同作用, 即NH2官能团促进有机配体到Fe中心的电子转移和Fe-O簇直接的激发, NH2修饰后的Fe基MOFs材料对CO2还原的光催化活性显著增强.
Zhang等[37]报道了Ru-MOF的花瓣状分级纳米结构, 其能够被用于可见光驱动的光催化还原CO2. 以三乙醇胺(TEOA)作为牺牲剂, 其在可见光照射下还原CO2生成HCOO-的速率可达77.2 μ mol· g-1· h-1, 并且不同尺寸下光催化还原CO2的活性顺序为纳米花> 微米级晶体> 块状晶体. 可见, 独特的花瓣状纳米结构显著提高了这种材料的光催化活性(比固体微晶的活性提高近150%). 此外, 花瓣状三维纳米结构也显著提高了Ru-MOF纳米片的稳定性和可回收性.
在有机配体修饰中, NH2官能团修饰可以显著提升MOFs的光催化效果. 主要原因是NH2基团对CO2有很高的亲和力, 有利于增强吸附能力. Dan-Hardi等[38]通过溶剂热法合成了MIL-125(Ti), 该材料较高的BET比表面积有利于CO2的吸附. 同时, 该MOF具有高密度且均匀分散的Ti活性位点. 在此研究基础上, Fu等[39]以2-氨基对苯二甲酸酯(ATA)为有机配体, 合成了NH2-MIL-125(Ti)并用于光催化还原CO2. 在可见光照射10 h后HCOO-的生成量达到8.14 μ mol. 这一方面是因为NH2的引入可以促进TiO5(OH)金属簇中的电子由O快速转移到Ti, 使NH2-MIL-125(Ti)在可见光区域具有良好的光吸收能力; 另一方面NH2还能够显著提高NH2-MIL-125(Ti)对CO2的吸附能力, 最终有利于光催化反应过程中CO2的吸附与活化.
与TiIV/TiⅢ 相比, ZrIV/ZrIII具有更低的氧化还原电位, 因此有利于光催化反应的发生. 基于以上考虑, Sun等[40]合成了NH2-UiO-66(Zr)并用于可见光下光催化还原CO2. 结果表明, NH2-UiO-66(Zr)的催化性能高于同等条件下的NH2-MIL-125(Ti), 这归因于光生电子可以有效地从ATA转移到Zr— O簇, 从而使Zr— O簇成为良好的光催化活性位点. 进一步用2, 5-二氨基对苯二甲酸(DTA)取代部分ATA配体, 获得混配NH2-UiO-66(Zr). 研究发现混配NH2-UiO-66(Zr)比纯NH2-UiO-66(Zr)的CO2转化率高50%. 这可能是因为混配NH2-UiO-66(Zr)具有较强的光吸收能力和较大的CO2吸附量, 使其光催化活性明显提高.
单一MOFs催化剂的催化中心通常是配位不饱和金属位点、 路易斯酸中心以及有机配体上的活性基团. 为了进一步提升单一MOFs在光催化还原CO2反应中的性能, 人们尝试对MOFs的金属节点或有机配体进行功能修饰, 或者将一些客体小分子引入到MOFs内作为电子受体, 以有效促进光生电子与空穴的分离和转移, 提高太阳能的利用率, 从而进一步提高光催化活性.
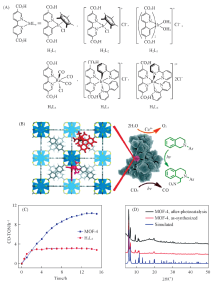
1.2.1 有机配体修饰的MOFs基光催化剂 通常, MOFs在水中稳定性较差. 基于Zr6O4(OH)4(CO2)12的次级结构单元和二羧酸桥联配体的UiO系列在水中非常稳定, Wang等[41]将具有二羧酸官能团的Ir, Re和Ru络合物H2L1-H2L6[化学结构见图2(A)]引入到高度稳定的UiO-67中. 其中将ReⅠ (CO)3(bpydc)Cl(H2L4)(bpydc=5, 5'-二羧酸-2, 2'-联吡啶)引入到UiO-67中得到的MOF-4可以作为可见光驱动下还原CO2的光催化剂[图2(B)]. 性能测试结果表明, 当反应时间增加到2 h后, MOF-4的催化活性开始高于H2L4, 并且能保持较好的稳定性[图2(C), (D)]. 这可能是因为长时间的反应下, H2L4自身容易发生光生电子与空穴的复合, 而MOF的特殊的晶体结构与孔道有利于光生电子与空穴的分离以及电子的迁移, 从而进一步提高光催化活性与转化效率.
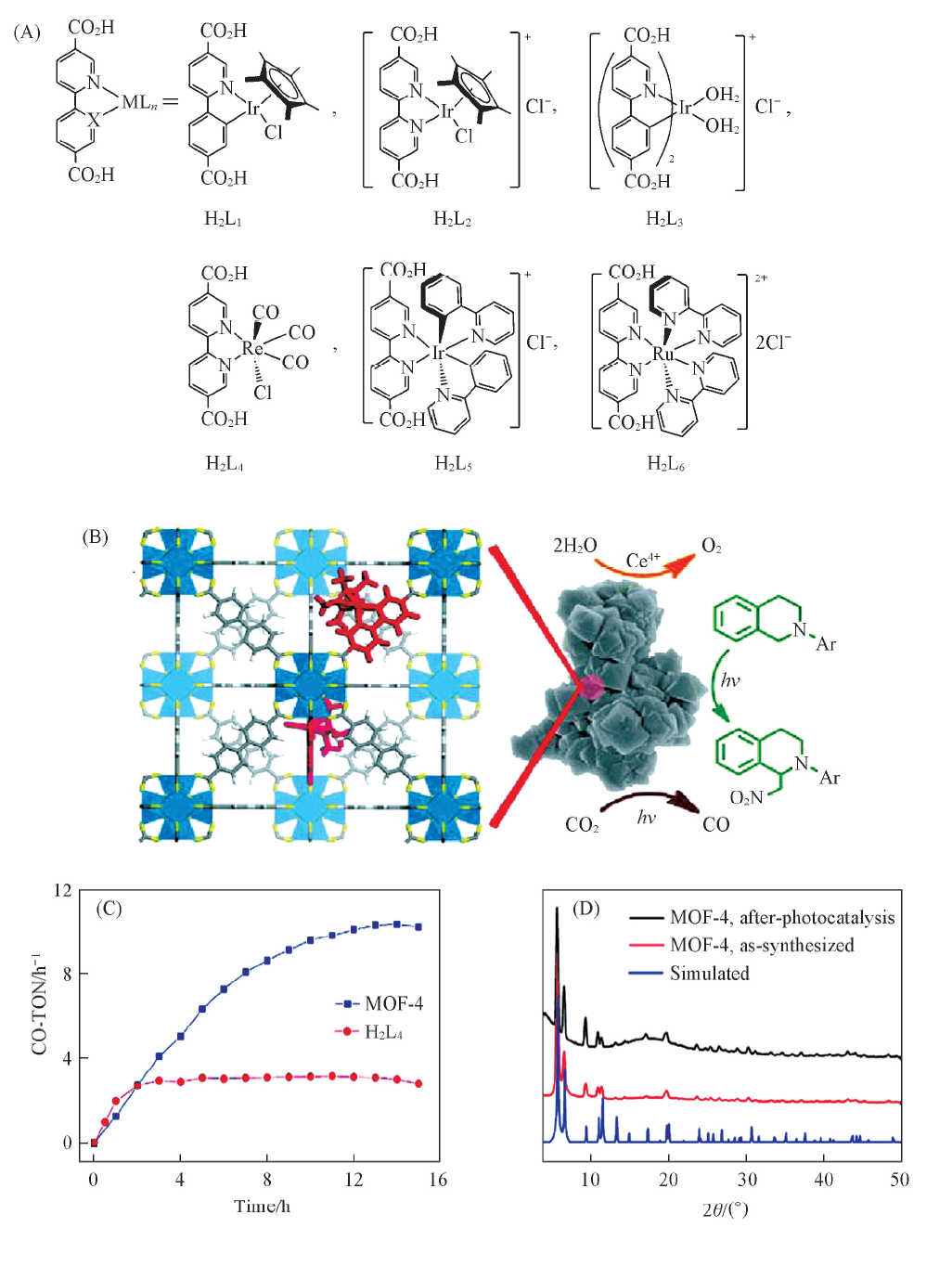 | Fig.2 Ligand structure of H2L1-H2L6(A), photocatalytic conversion schematic(B), plots of CO-TON versus time(C) and PXRD patterns of the catalysts(D)[41] Copyright 2011, American Chemical Society. |
Choi等[42]将光催化剂ReⅠ (CO)3(bpydc)Cl(记为ReTC)共价连接至UiO-67得到Ren-MOF(n为ReTC在MOF孔隙中的密度), 然后进一步将该MOF与立方纳米银复合得到Ag⊂Ren-MOF, 从而使光催化CO2转化的活性显著提高[图3(A)]. PXRD测试结果表明, 在Ren-MOF中引入不同密度的ReTC时保留了单晶Re3-MOF结构[图3(B)]. 通过对Ren-MOF光催化转化CO2的过程进行研究[图3(C)], 发现Re3-MOF的催化活性最高. 此外, 在可见光照射下, Ag⊂Re3-MOF的活性比Re3-MOF提高了5倍, CO2转化生成CO的效率提高了7倍, 稳定性也极大提高. 这主要是因为一方面MOF具有多孔性以及较大的CO2吸附量, 有利于催化还原反应的发生; 另一方面贵金属具有较宽的光吸收范围, 更易快速产生光生电子. 同时, 由于ReTC与MOF之间较强的共价键作用, 使其稳定性进一步提高.
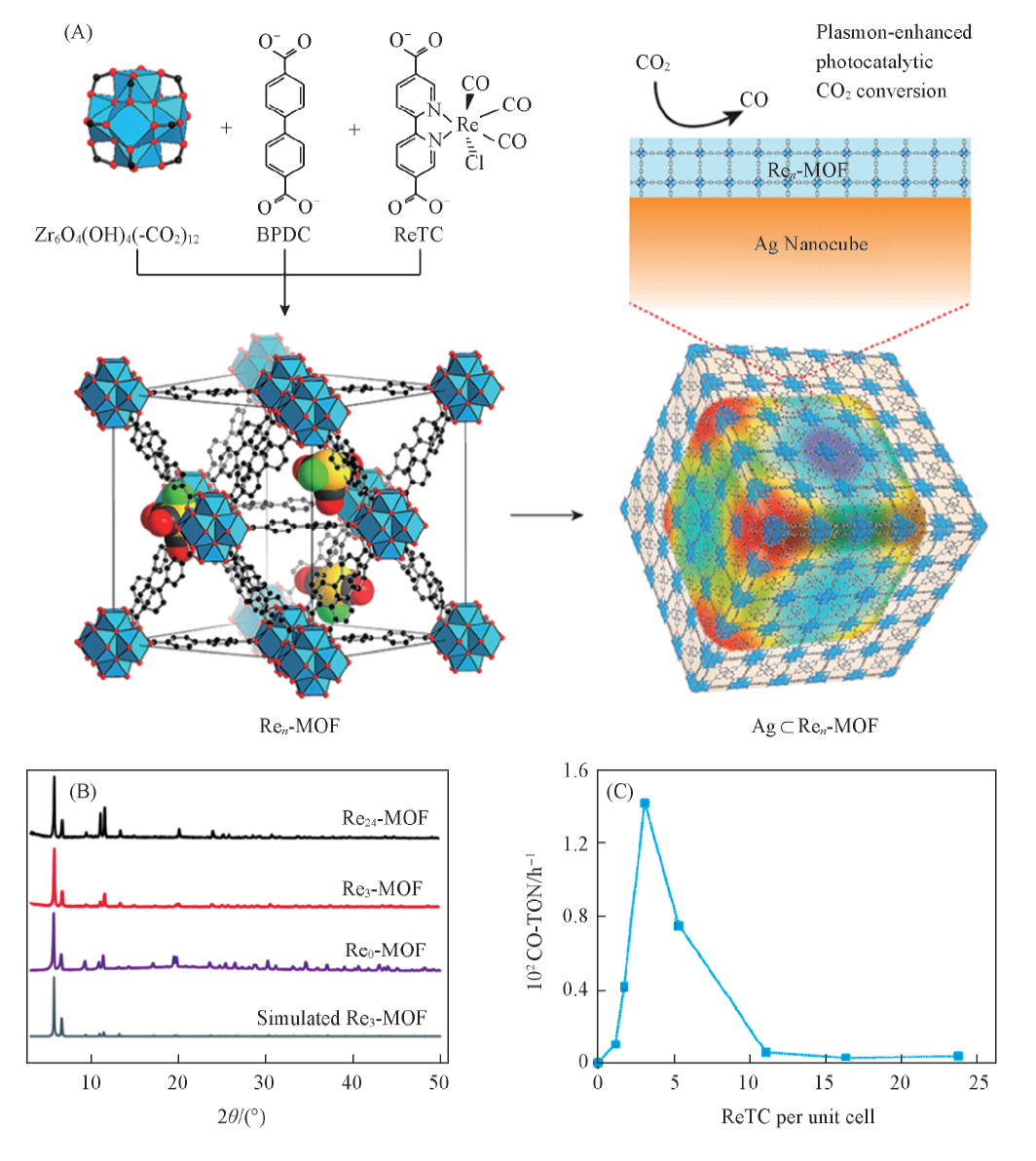 | Fig.3 Structures of Ren-MOF and Ag⊂Ren-MOF based catalysts(A), PXRD of Ren-MOFs(B) and the photocatalytic activity of Ren-MOF(C)[42] Copyright 2017, American Chemical Society. |
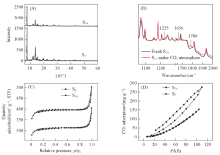
1.2.2 金属离子/离子簇修饰的MOFs基光催化剂 Liu等[43]将Al-卟啉MOF中引入Cu2+, 研究了其光催化还原CO2的性能(引入Cu2+的记为SCu, 未引入Cu2+的记为SP). 结果表明, 引入Cu2+前后晶体结构基本没有发生变化[图4(A)]. 原位FTIR光谱显示SCu的CO2吸附等温线发生滞后, 这可能是因为CO2在Cu2+活性位点上发生了化学吸附[图4(B)]. 此外, SP和SCu的BET比表面积分别为1187和932 cm2/g[图4(C)]. SCu较小的BET比表面积可能是由于将Cu2+引入到卟啉中阻塞了SP中的卟啉孔. 尽管SCu的BET比表面积降低了20%, 但其CO2吸附能力高出80%[图4(D)]. 光催化实验结果表明, SCu作光催化剂时CH3OH的生成速率是SP作光催化剂时的7倍. SCu催化性能提高主要是因为Cu2+对CO2的化学吸附和活化, 在化学吸附的作用下稳定的线性CO2分子发生弯曲, 从而使光催化还原的活化能下降, 催化活性得到提升.
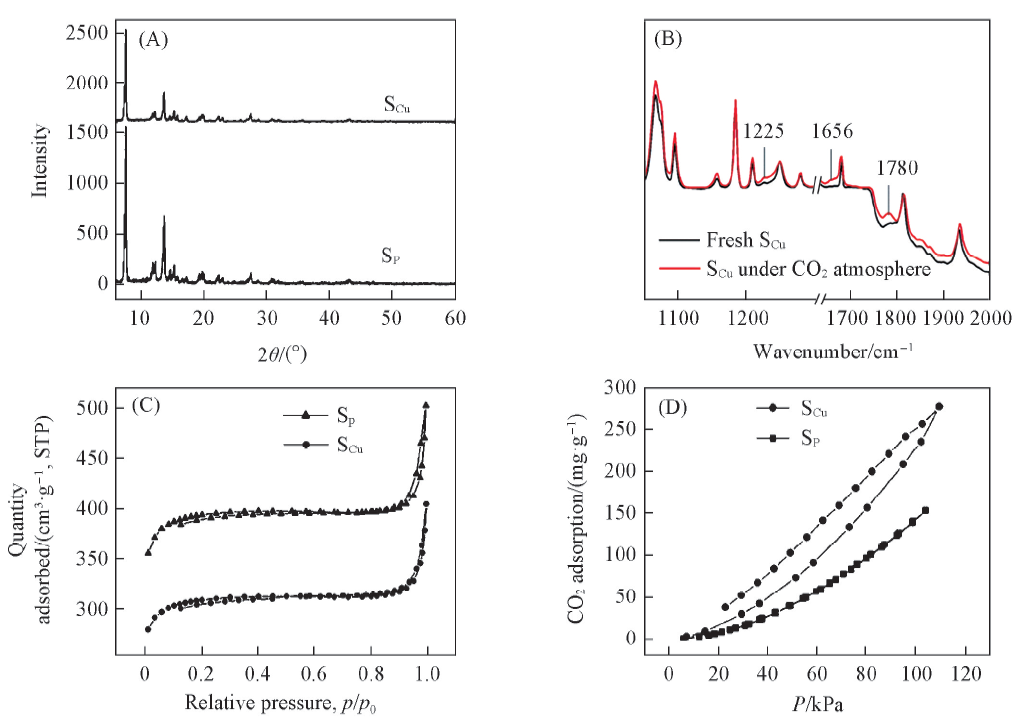 | Fig.4 XRD patterns(A) and FTIR spectra(B), N2(C) and CO2(D) adsorption and desorption isotherms of SCu and Copyright 2013, American Chemical Society. |
研究发现, 将具有光氧化还原活性的材料作为客体引入到MOFs的孔隙中可以有效提升催化性能. Fei等[44]将Mn(bpydc)-(CO)3Br引入到高度稳定的UiO-67中提升了其CO2还原的光催化性能. 在作为氧化还原光敏剂的[Ru(dmb)3]2+(dmb=4, 4'-二甲基-2, 2'-联吡啶)和作为牺牲还原剂的1-苄基-1, 4-二氢烟酰胺(BNAH)的辅助下, 该材料能够光催化还原CO2生成HCOO-. 在可见光照射下, UiO-67-Mn(bpy)(CO)3Br作为光催化剂可高选择性生成HCOO-. Mn-UiO-67催化活性提高首先是由于CO2的吸附量提高; 其次, MOF提供了分散的活性位点和稳定的框架结构, 可有效抑制还原性Mn配合物的二聚, 增强了稳定性; 最后, 引进光敏剂有助于光生电子的迁移.
Lee等[45]以UiO-66(锆1, 4-羧基苯)为前体, 用2, 3-二羟基对苯二甲酸(H2catbdc)通过后合成交换的方法获得了UiO-66-CAT, 然后分别将2种金属离子Cr3+和Ga3+结合到CAT位点上得到UiO-66-CrCAT和UiO-66-GaCAT. CAT配体可以有效增强可见光的吸收, 而Cr3+和Ga3+可以促进MOF内快速电子传输. 在TEOA和BNAH存在下, UiO-66-CrCAT和UiO-66-GaCAT的转化数(TON)值分别为11.22± 0.37和6.14± 0.22, 可见光照射6 h后催化还原CO2生成HCOOH的量分别为(51.73± 2.64)和(28.78± 2.52) μ mol. UiO-66-CrCAT的活性约是UiO-66-GaCAT的2倍, 这主要归因于Cr3+比Ga3+更有利于电子的快速传递. 同时, Cr衍生物由于其开放的壳体电子结构比Ga衍生物显示出更高的还原效率.
除了直接用作光催化剂之外, 由于MOFs的电子传导性差, 以及所有催化活性位点对反应物不可接近, MOFs在水中和紫外光照射下的稳定性需要进一步提高. 为了取得更好的催化效果, 有必要使用MOF来构建更复杂的材料, 以在一种材料中同时解决CO2吸附和还原问题. 这种复合结构可以促进光生电子的分离和转移, 从而进一步提高光催化活性. 其中MOFs可以促进催化反应的动力学过程和增强CO2吸附, 而其它组分(如染料、 半导体等)在纳米复合材料中作为光吸收部分可以形成光生载流子[46].
2.1.1 MOFs/无机半导体复合光催化剂 由于无机半导体的光激发特性和MOFs高的CO2吸附能力产生的协同效应, 将MOFs材料与无机催化剂复合制备杂化催化剂可以有效提高催化活性. 其中半导体TiO2作为光催化剂已经被广泛研究[4749], 而TiO2/MOFs复合材料最近几年也被进一步研究. Yan等[50]将不同量的TiO2负载在Co-ZIF-9上构筑Co-ZIF-9/TiO2纳米结构的复合材料(ZIFx/T, x为复合材料中Co-ZIF-9的质量比). 结果表明, ZIF0.03/T具有最佳的CO2催化转化效率, 在光照10 h后的产率约为纯TiO2催化剂的2.1倍. 在CO2饱和溶液中的线性扫描伏安测试进一步揭示了Co-ZIF-9可以有效活化CO2并且降低ZIFx/T(x≤ 0.10)的CO2还原起始电位. 此外, 光致发光光谱表明, 通过原位合成方法制备的ZIFx/T复合材料具有更高的电荷分离效率. 因此, 更好的CO2吸附能力和电荷分离速率都有利于ZIFx/T纳米结构在光催化转化中的高活性.
Maina等[51]设计了基于膜反应器的催化体系, 通过快速热沉积(RTD)方法, 实现了TiO2和Cu2+掺杂的TiO2纳米粒子(Cu-TiO2)在ZIF-8膜内的可控封装[图5(A)]. 在紫外光照射下, Cu-TiO2/ZIF-8杂化膜显示出很强的光催化活性. 结果表明, 与单独使用原始ZIF-8膜产生的量相比, CO和CH3OH的产率分别增加188%和50%[图5(B)]. 进一步研究表明, 光催化还原CO2生成CH3OH和CO的产率取决于负载到MOF膜上的Cu-TiO2纳米颗粒的含量[图5(C)]. 当Cu-TiO2纳米颗粒的负载量为7 μ g时, Cu-TiO2/ZIF-8具有最好的催化效率, 比原始ZIF-8膜的CO和CH3OH产率分别提高了约233%和约70%. 产物产率的急剧增大体现了半导体纳米粒子在光照射下产生光激发电子的能力和MOF的高CO2吸附能力所产生的协同效应.
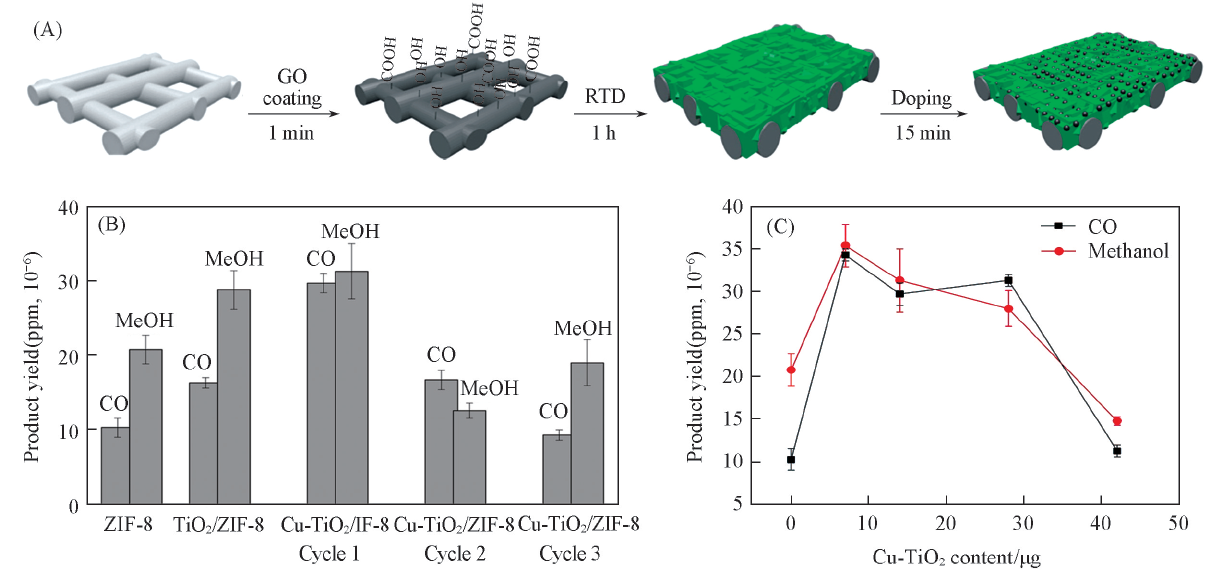 | Fig.5 Fabrication of Cu-TiO2/ZIF-8 membranes(A), effect of membrane composition(B) and Cu-TiO2 nanoparticles loading on the product yields(C)[51] Copyright 2017, American Chemical Society. |
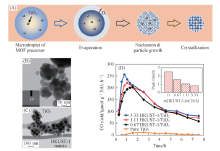
除了在MOFs表面上涂覆半导体的方法外, He等[52]提出利用气溶胶技术进行复合物的制备, 以改善传统方法中的一些缺陷, 如半导体可能会阻挡气体吸附位点和传输通道, 从而限制MOFs的气体吸附能力. 他们利用气溶胶技术将铜基金属有机框架(HKUST-1)和TiO2复合, 得到了复合材料. 如图6(A)所示, 将前体溶液通过雾化器雾化成微滴, 并通过载气在预定温度下携带, 其中微滴经历溶剂蒸发、 成核和蒸发驱动的自组装过程. HKUST-1/TiO2经进一步干燥后形成颗粒[图6(B), (C)]. 结果表明, 纯TiO2在照射2.53 h后仅能产生11.48 μ mol· g-1· h-1的CO. 随着加入HKUST-1量的增加, CO生成速率逐渐增大. 当HKUST-1/TiO2的摩尔比增加到3.33时, CO2的转化效率增加了20倍以上[图6(D)]. 催化性能显著提升是由于一方面合成的HKUST-1基纳米材料结晶度好, 比表面积大, 孔隙率高且光吸收范围较宽; 另一方面将TiO2嵌入HKUST-1基质后, 增强了载流子的分离效率, 进而提高了CO2光还原效率, 同时增强了反应物的吸附量和HKUST-1的稳定性.
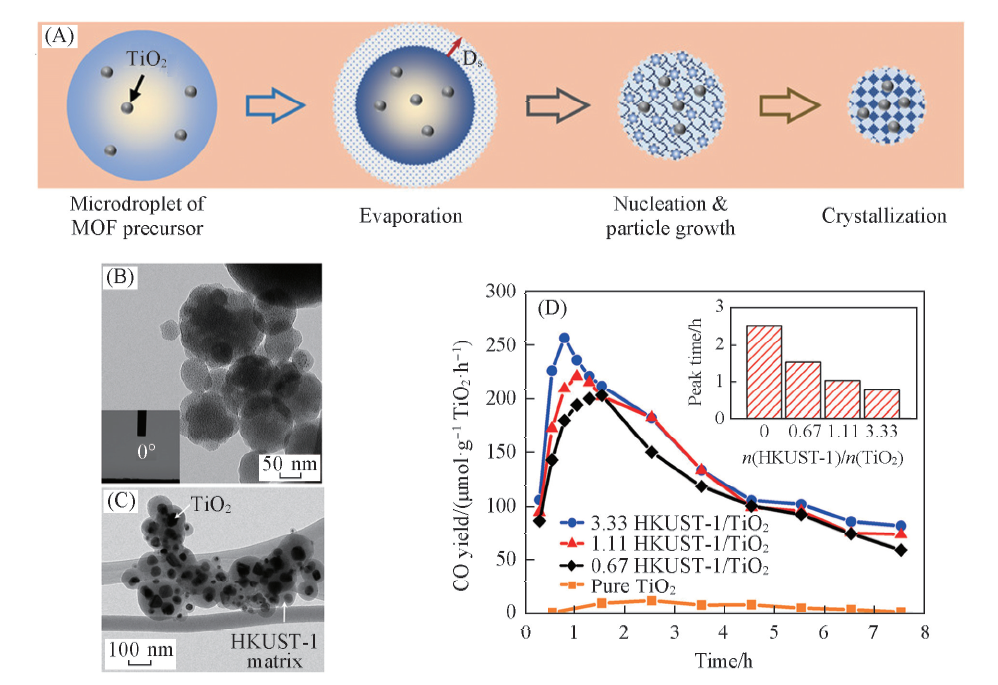 | Fig.6 HKUST-1 and HKUST-1/TiO2 formation steps inside a microdroplet(A), TEM images of as-synthesized HKUST-1(B) and 33.3 HKUST-1/TiO2(C) at 300 ℃ and CO2 photoreduction performance of TiO2 and HKUST-1/TiO2 composites(D)[52] Inset of (B): the image of the contact angle measurement of HKUST-1 surface. Copyright 2017, American Chemical Society. |
Liu等[53]首先通过水热合成法合成了Zn2GeO4, 再采用原位生长法将ZIF-8生长在Zn2GeO4半导体表面, 获得了Zn2GeO4/ZIF-8杂化纳米棒. 测试结果表明, 相比于纯Zn2GeO4, 其CO2吸收量提高了约7倍, 主要原因是表面修饰的ZIF-8具有优异的CO2吸附能力. 其中Zn2GeO4/ZIF-8复合催化剂催化CO2生成CH3OH的速率最高, 为0.22 μ mol· g-1· h-1. 相比于Zn2GeO4纳米棒光催化剂, 在可见光照射10 h后Zn2GeO4/ZIF-8杂化纳米棒催化CO2还原为CH3OH的产量提高了62%. 主要原因是引入ZIF-8能有效吸附水溶液中溶解的CO2, 继而促进半导体将含水的CO2还原为CH3OH, 并且Zn2GeO4/ZIF-8的光吸收也优于Zn2GeO4.
在非金属半导体光催化剂的研究中, 石墨相氮化碳(g-C3N4)因具有较高的热稳定性和化学稳定性、 适宜的能带结构和良好的可见光响应能力而被引入到各种光催化应用中[5456]. 但g-C3N4的光催化剂电荷分离能力和CO2吸附能力仍有待进一步提高. Liu等[57]首先制备了多孔g-C3N4纳米管, 再用适量的ZIF-8修饰制备了管状g-C3N4(TCN). 块状g-C3N4(BCN)表现出相对高的CO2吸附性能. 在BCN或TCN修饰了ZIF-8后, CO2还原性能明显增强. 当修饰的ZIF-8与g-C3N4(TCN)的质量比为1∶ 8(即样品TCNZ8)时, 光催化的CH3OH生成速率最高, 达到0.75 μ mol· g-1· h-1. 催化活性的显著提升归因于g-C3N4较强的光捕获能力和电荷分离效率与ZIF-8较高CO2吸附能力的协同作用.
2.1.2 无机半导体@MOFs复合光催化剂 Li等[58]将TiO2纳米粒子沉积在Cu3(BTC)2表面获得了具有核壳结构的Cu3(BTC)2@TiO2复合光催化剂, 其在紫外光照射下可催化还原CO2为CH4. 结果表明, 紫外光照射4 h, 以单一TiO2作为光催化剂时, 生成产物为CH4和H2; 单一Cu3(BTC)2因为其共轭结构不利于电荷分离, 因而不具有光催化活性; 相比之下, 具有核壳结构Cu3(BTC)2@TiO2作为光催化剂时, CH4的生成速率约为单一TiO2光催化剂时的5倍, 同时没有检测到H2生成. 因此, Cu3(BTC)2@TiO2在光催化CO2转化为CH4的过程中具有优异的转化效率和选择性. 此外, Cu3(BTC)2@TiO2还具有很好的稳定性. 这可能是由于该复合材料在光照条件下, 外层的TiO2被光激发产生电子-空穴对, 电子可以快速转移到Cu3(BTC)2上; 同时, Cu3(BTC)2具有较高的CO2吸附能力, CO2吸附在Cu金属位点上进行还原反应.
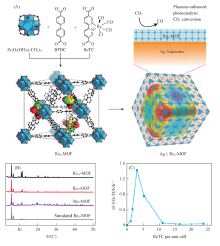
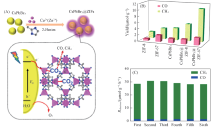
Kong等[59]采用原位合成的方法制备了具有增强活性的CO2还原光催化剂CsPbBr3@ZIFs复合材料[图7(A)]. 复合催化剂CsPbBr3 @ZIF-8和CsPbBr3@ZIF-67的电子消耗率分别为15.498和29.630 μ mol· g-1· h-1, 表现出较强的CO2还原活性, 分别是纯CsPbBr3的1.39和2.66倍. CsPbBr3和CsPbBr3@ZIFs的光催化CO2还原性能表明ZIF涂层大大提高了CsPbBr3的催化活性, 特别是对于CsPbBr3@ZIF-67[图7(B)]. 此外, 对CsPbBr3@ZIF进行了6次循环实验, 发现电子消耗率没有明显的下降, 表明其具有良好的稳定性[图7(C)]. CsPbBr3和ZIF涂层的协同作用改善了CsPbBr3的水稳定性, 提高了CO2捕获能力和电荷分离效率, 这有助于提高转化效率. 而且, ZIF-67中的催化活性中心Co能进一步加速电荷分离的过程, 活化CO2分子, 提高CO2还原的催化活性.
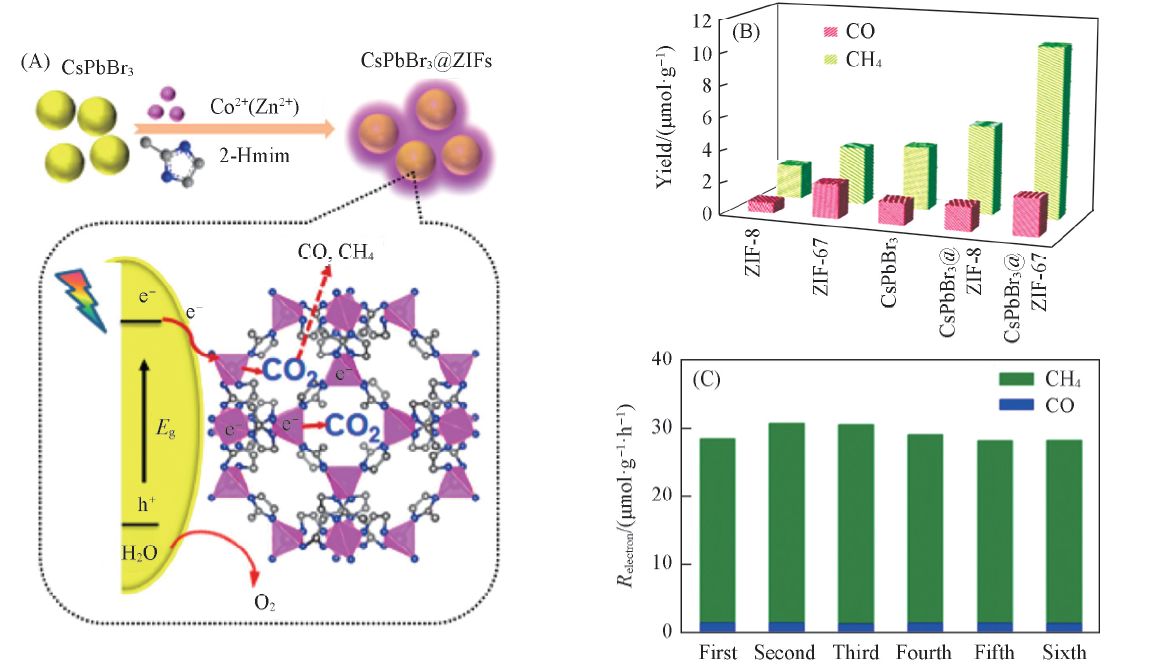 | Fig.7 Schematic illustration of the fabrication process and CO2 photoreduction process of CsPbBr3/ZIFs(A) and photocatalytic CO2 reduction performances of CsPbBr3 and CsPbBr3@ZIFs(B, C)[59] Copyright 2018, American Chemical Society. |
2.1.3 MOFs@无机半导体核壳光催化剂 Su等[60]采用溶剂热法制备了一系列不同UiO-66-NH2含量的Cd0.2Zn0.8S@UiO-66-NH2复合材料, 并用于光催化还原CO2为CH3OH. 结果表明, 单一UiO-66-NH2不能光催化还原CO2; CdxZ
Sun等[61]使用Ru羰基配合物作为有机配体来制备MOFs催化剂, 并用于光催化还原CO2为HCOO-和CO. 尽管MOF-253-Ru(CO)2Cl2对CO2还原具有光催化活性, 但其性能并不显著, 因此利用光敏性材料可以提高光催化剂的光吸收能力, 进而提高催化还原效率. 研究发现, 当催化剂中Ru(bpy)2Cl2/MOF-253-Ru(CO)2Cl2的摩尔比为1∶ 1时, 与未敏化的催化剂相比, 光照8 h后产生的CO量没有太大变化, 但HCOO-的量提高了约12倍. 这说明MOF-253不仅可以作为构建光催化剂MOF-253-Ru(CO)2Cl2的主体, 而且还可以作为构建复合光催化体系的平台, 从而促进光敏剂和表面构造的光催化剂之间的电荷转移. 然而Ru(CO)2Cl2量的不断增加可能导致反应活性降低, 这可能是因为含量过多的Ru(CO)2Cl2堵塞了MOF-253的孔结构, 从而使反应物与光催化剂无法接触.
Qin等[62]以ZIF-67作为助催化剂, 通过与染料光敏剂[Ru(bpy)3]Cl2· 6H2O(bpy=2, 2'-联吡啶)配合, 在可见光照射下光催化还原CO2为CO. 实验结果表明, 助催化剂ZIF-67在温和反应条件下具有一定的稳定性. 同时, 当催化体系分散8 mg光敏剂[Ru(bpy)3]Cl2· 6H2O时, 加入0.1 mg ZIF-67后, CO的产率提高到37.4 μ mol/30 min, 明显高于其它类型的MOFs. 而且通过与其它MOFs相比, 发现ZIF-67的钴活性位点对于促进电子传输起到了关键作用. 因此, 较高的催化活性一方面是因为ZIF-67较高的表面积和微孔性能, 为表面电荷载体的迁移和捕获提供了有效的传输途径, 便于加速将吸附的CO2还原为CO; 另一方面光敏剂的加入增强了光激发能力, 从而进一步提高光催化还原性能.
此外, Shi等[63]通过静电自组装构筑制备了UiO-66/氮化碳纳米片(CNNS)非均相光催化剂. 其在可见光照射下, 可以将CO2催化还原为CO. 单一UiO-66不具有催化还原CO2的能力, 而加入CNNS形成复合材料后有利于分离光激发的电子-空穴对, 从而使CO的生成速率明显提高, 并且比单一CNNS高1.6倍. Zhang等[64]报道了单原子Co负载的Zr-卟啉MOF(MOF-525-Co), 其暴露的活性位点不仅能够有效吸附CO2, 而且可以分散活性位点. 以TEOA作为牺牲剂, MOF-525-Co在可见光照射下可以高效催化还原CO2为CO和CH4. 与Zn-MOF-525和MOF-525相比, MOF-525-Co显示出最高的催化活性和CO2吸附量. 金属化后的MOFs具有较强的电荷分离能力, 能量转化效率明显提高, 而钴金属化后催化性能最高主要是因为MOF-525引入单原子Co后可以显著提高卟啉配体中的电子-空穴分离效率, 同时, 光生电子从卟啉中心快速迁移到催化剂表面, 从而获得了具有长寿命的电子, 进而有效活化了吸附在Co中心上的CO2.
MOFs基材料在光催化还原CO2方面展现了显著优势, 已成为近年来光催化领域的一类新型材料. 尽管如此, 在该领域中MOFs的发展依然面临许多问题. 首先, 已报道的MOFs基材料光催化CO2的效率依然较低, 部分原因是由于部分MOFs的电子传输能力较差以及对太阳能的吸收效率较低; 其次, 大多数MOFs不如半导体光催化剂稳定, 特别是在水中或在紫外光照射下结构稳定性下降, 最终导致催化剂的寿命下降; 第三, MOFs光催化CO2还原机理研究尚少, 特别是当前人们对催化反应路径的认识依然模糊. 此外, 大多数报道的光催化CO2还原反应在有机溶剂中进行, 需要额外加入牺牲剂, 而未来用于CO2催化还原的材料应该是经济环保的. 后期MOFs光催化CO2转化过程的关键包括在MOFs中设计结构缺陷、 金属簇和有机金属接头的Lewis酸位点以及助催化剂的功能化. 同时, 设计合成高活性、 高稳定性以及可循环利用的MOFs光催化剂用于还原CO2还需要进行深入研究.
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|
| [20] |
|
| [21] |
|
| [22] |
|
| [23] |
|
| [24] |
|
| [25] |
|
| [26] |
|
| [27] |
|
| [28] |
|
| [29] |
|
| [30] |
|
| [31] |
|
| [32] |
|
| [33] |
|
| [34] |
|
| [35] |
|
| [36] |
|
| [37] |
|
| [38] |
|
| [39] |
|
| [40] |
|
| [41] |
|
| [42] |
|
| [43] |
|
| [44] |
|
| [45] |
|
| [46] |
|
| [47] |
|
| [48] |
|
| [49] |
|
| [50] |
|
| [51] |
|
| [52] |
|
| [53] |
|
| [54] |
|
| [55] |
|
| [56] |
|
| [57] |
|
| [58] |
|
| [59] |
|
| [60] |
|
| [61] |
|
| [62] |
|
| [63] |
|
| [64] |
|