






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
聚(L-谷氨酸)水凝胶介导羟基磷灰石的生物矿化
[颜世峰 , 王卫东, 任婕, 滕畅畅, 尹静波]
, 王卫东, 任婕, 滕畅畅, 尹静波]
, 王卫东, 任婕, 滕畅畅, 尹静波]
|
|


联系人简介: 颜世峰, 男, 博士, 教授, 主要从事医用高分子材料方面的研究. E-mail: yansf@staff.shu.edu.cn;尹静波, 女, 博士, 教授, 主要从事医用高分子材料方面的研究. E-mail: jbyin@oa.shu.edu.cn
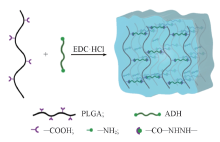
以1-乙基-3-(3-二甲基氨基丙基)碳二亚胺盐酸盐(EDC·HCl)为羧基活化剂, 己二酸二酰肼(ADH)为交联剂, 制备了生物活性聚(L-谷氨酸)(PLGA)水凝胶. 通过X射线衍射和扫描电子显微镜等表征了在不同浓度模拟体液(SBF)中羟基磷灰石(HA)的形成和生长. PLGA水凝胶的表面和内部均可观察到HA的形成和生长. 同时探讨了PLGA水凝胶矿化前后的力学性能. 将矿化前后PLGA水凝胶用于脂肪干细胞(ASCs)的培养, 研究其细胞相容性.
Bioactive poly(L-glutamic acid)(PLGA) hydrogels were prepared by using adipic acid dihydrazide(ADH) as a crosslinking agent. The formation and growth of hydroxyapatite(HA) in the simulated body fluid(SBF) of different concentrations were characterized by various techniques. PLGA is a kind of anionic poly(amino acid) with abundant carboxylic groups, which favors the enrichment of calcium ions and formation of HA. The growth of HA with soaking time was detected at the surface and interior of the PLGA hydrogels. The mechanical properties of the PLGA hydrogels before and after mineralization were studied in dynamic rheology experiments. Being cytocompatible, the mineralized PLGA hydrogels were successfully applied for cultivation of adipose-derived stem cells(ASCs). These results presented a method of preparing HA/PLGA composite hydrogels, which might be used as a promising biomaterial for bone tissue engineering.
在骨组织工程中, 理想的支架材料应具有诱导钙化能力[1]. 天然骨在结构上是由胶原(一种基于蛋白质的水凝胶模板)和羟基磷灰石(HA)组成的有机/无机复合材料[2]. HA具有良好的生物相容性和骨传导性[3], 但HA硬而脆的特性限制了其在骨修复领域的应用. 将HA与弹性聚合物水凝胶网络相结合可制备柔性生物活性复合材料, 其低刚度、 抗外力、 高断裂韧性[4]以及易成型的优点而备受关注[5]. 此外, 在水凝胶的制备过程中, 可通过引入极性配体(如羧基), 来模仿调节矿物质生长的骨基质酸性蛋白[6], 模拟骨的形成过程.
制备聚合物/HA复合材料的方法通常有3种: 与HA纳米颗粒直接混合[7]、 聚合物存在下HA沉淀[8]和仿生矿化[9]. 虽然前2种方法简单且成本低, 但难以实现表面化学性质和形貌的调控[10, 11]. 而生物矿化方法, 即模仿生物系统的矿化过程, 是一种简单有效的模拟复杂形状类骨结构的方法. 通常, 用于生物矿化的聚合物模板包括天然生物大分子, 如胶原蛋白、 明胶、 多糖[12], 以及合成聚合物[13](如聚丙烯酸[14]). 受天然骨组织形成过程中蛋白质调控矿化的启发, 在蛋白质及其类似物存在下调控HA生长是最常用的一种生物矿化研究模型[15]. 骨唾液蛋白(BSP)是一种骨特异性糖蛋白, 含有磷酸丝氨酸及硫酸酪氨酸残基且富含谷氨酸残基, 这些氨基酸残基可以作为HA异相成核的引发剂[16, 17, 18]. BSP中谷氨酸残基的化学修饰会导致HA的成核能力降低, 表明BSP中富含的谷氨酸序列在成核中起重要作用[17, 19]. 基于胶原蛋白的矿化过程已被广泛研究[20, 21], 但由于天然来源的胶原提取过程复杂, 并且可能具有免疫原性反应, 加之材料成本高、 强度低, 在骨组织支架中胶原基材料的使用受到限制[11, 22].
制备聚合物/HA复合材料的方法通常有3种: 与HA纳米颗粒直接混合[7]、 聚合物存在下HA沉淀[8]和仿生矿化[9]. 虽然前2种方法简单且成本低, 但难以实现表面化学性质和形貌的调控[10, 11]. 而生物矿化方法, 即模仿生物系统的矿化过程, 是一种简单有效的模拟复杂形状类骨结构的方法. 通常, 用于生物矿化的聚合物模板包括天然生物大分子, 如胶原蛋白、 明胶、 多糖[12], 以及合成聚合物[13](如聚丙烯酸[14]). 受天然骨组织形成过程中蛋白质调控矿化的启发, 在蛋白质及其类似物存在下调控HA生长是最常用的一种生物矿化研究模型[15]. 骨唾液蛋白(BSP)是一种骨特异性糖蛋白, 含有磷酸丝氨酸及硫酸酪氨酸残基且富含谷氨酸残基, 这些氨基酸残基可以作为HA异相成核的引发剂[16, 17, 18]. BSP中谷氨酸残基的化学修饰会导致HA的成核能力降低, 表明BSP中富含的谷氨酸序列在成核中起重要作用[17, 19]. 基于胶原蛋白的矿化过程已被广泛研究[20, 21], 但由于天然来源的胶原提取过程复杂, 并且可能具有免疫原性反应, 加之材料成本高、 强度低, 在骨组织支架中胶原基材料的使用受到限制[11, 22].
考虑到BSP中谷氨酸残基能够诱导矿化, 本文研究了聚(L-谷氨酸)(PLGA)水凝胶的生物矿化. PLGA是一种具有良好生物相容性的合成多肽, 由天然存在的L-谷氨酸通过酰胺键连接而成, 具有亲水性、 生物降解性、 非抗原性和免疫原性的优点. PLGA因其分子链侧链上具有羧基的残基具有诱导HA形成的能力[23]. 本文以己二酸二酰肼(ADH)为交联剂, 通过化学交联构建PLGA水凝胶, 评估了PLGA水凝胶在SBF中的矿化能力, 并提出可能的矿化机理.
聚(L-谷氨酸)(PLGA, Mη =5.0× 104)参照文献[24]方法制备. 己二酸二酰肼(ADH)和1-乙基-3-(3-二甲基氨基丙基)碳二亚胺盐酸盐(EDC· HCl)购自上海达瑞化学品公司.
1.2.1 PLGA水凝胶的制备 配制质量分数为6%的PLGA水溶液, 加入ADH, 利用NaOH或HCl调节溶液的pH至4.5, 搅拌2 h后加入EDC, 控制ADH/EDC/PLGA中— COOH摩尔比为0.325: 1: 1.将溶液快速混合均匀, 静置反应4 h后形成透明水凝胶.
1.2.2 PLGA水凝胶的仿生矿化 将PLGA水凝胶浸泡于模拟体液(1.5SBF和1.0SBF)中以诱导HA的形成. 1.5SBF和1.0SBF是按照模拟血浆中的离子浓度而配制的盐溶液, 主要成分如表1所示. 其中1.5SBF的离子浓度是1.0SBF的1.5倍, 可加速HA的生长[25]. 浸泡2, 6, 10, 14, 21和28 d后, 取出水凝胶, 用蒸馏水洗涤3次, 利用滤纸吸去表面残留水分, 将水凝胶置于液氮中快速冷冻, 而后置于冷冻干燥机内干燥48 h.
| Table 1 Comparation of the ion concentration(mmol/L) in human plasma, 1.0SBF and 1.5SBF |
1.2.3 测试与表征 利用X射线衍射仪(XRD, D/MAX2550, Rigaku, 日本)和傅里叶变换红外光谱仪(FTIR, AVATAR 370, Nicolet, 美国)表征冻干水凝胶以确认样品的矿化成分. 将材料切开, 喷金, 通过扫描电子显微镜(SEM, JSM-6700F, 日本电子, 日本)观测水凝胶矿化后的表面形貌, 通过X射线能谱(EDS)表征样品表面元素分析. 称量一定量的冻干水凝胶, 用热重分析仪(Q500, TA Instruments, 美国)评估SBF矿化不同时间HA的含量. 将水凝胶从SBF溶液中取出, 通过D-HR3旋转流变仪(D-HR3, TA Instruments, 美国)检测水凝胶矿化前后流变学性能.
参照文献[26, 27]方法从新西兰白兔后颈处提取兔脂肪干细胞(ASCs). 将冷冻干燥的PLGA水凝胶和经1.5SBF浸泡14 d的PLGA水凝胶置于75%乙醇中1 h后灭菌后, 将细胞接种到水凝胶支架上, 细胞接种密度为5× 107 Cell/mL[27]. 经1, 7, 14 d培养后将细胞/水凝胶复合物取出, 经2.5%(体积分数)戊二醛固定液固定过夜, 分别以25%, 70%, 80%, 95%的乙醇溶液梯度脱水, 每个梯度5 min; 再以梯度乙醇/六甲基二硅胺烷混合溶液(体积比为3: 1, 1: 1, 1: 3, 0: 1)逐级置换乙醇, 每级10 min, 干燥, 切除表层暴露截面, 喷金, 采用SEM观察细胞黏附和细胞外基质沉积情况. 使用活-死细胞染色试剂盒(Invitrogen)研究材料的生物相容性, 并采用共聚焦激光显微镜(CLSM, FV-1000, Olympus, 日本)观察细胞的存活情况.
PLGA可通过含有2个酰肼基团的低分子量交联剂ADH交联. 将EDC作为羧基的活化剂, 形成具有氨基反应活性的O-酰基脲中间体, 然后与酰肼基团发生缩合反应, 形成酰腙化学键[28], 以实现羧基和氨基之间的“ 零长” 交联[29]. 如Scheme 1所示, PLGA侧链羧基与ADH通过酰胺键交联形成三维网络结构. 据文献[30]报道, 在pH=3.54.5范围内, EDC介导的缩合反应的效率较高. 但当 pH< 4.4(PLGA的pKa=4.4)时, 水相中的PLGA链易蜷曲并沉淀[31], 因此反应过程中将溶液的pH设为4.5. 图1为加入EDC前后的PLGA/ADH混合物溶液形态. 在未加入EDC时, PLGA不能直接通过ADH交联形成水凝胶. 加入EDC后, 可得到透明的PLGA水凝胶, 表明活化的PLGA可与ADH反应形成三维聚合物网络. 水凝胶在被镊子夹起后仍能保持原始形状, 表明其良好的机械强度.
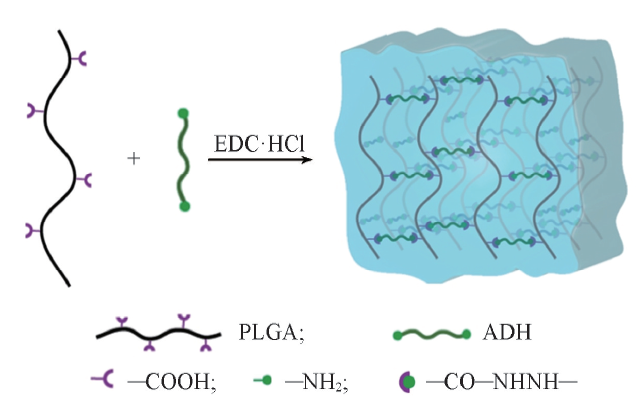 | Scheme 1 Schematic of PLGA crosslinking |
 | Fig.1 Photographs of PLGA/ADH solution before(A) and after addition of EDC(B— D) (B) Investion; (C) lifting up; (D) picking up. |
矿化PLGA水凝胶的XRD谱如图2(A)所示. 对于PLGA水凝胶, 其XRD图谱仅在2θ ≈ 21° 处出现一个宽峰, 表明其为无定形结构. PLGA水凝胶在1.5SBF中浸泡2 d后, 其XRD图谱中并未显示HA晶体的特征峰. 浸泡6 d后开始观察到HA的特征峰, 其中最强的峰位于2θ =32° 处, 这归因于HA的(211), (112)和(300)晶面衍射峰的叠加[32]. 随着浸泡时间的增加, 在2θ =39° , 46° 和49° 处出现新的特征峰[24], 并且所有特征峰的强度均随着浸泡时间增加而逐渐增加, 这表明PLGA水凝胶基质中HA的成核和生长. 同时, PLGA水凝胶在2θ =21° 处的特征峰强度减弱, 这归因于PLGA/HA复合材料中HA含量的逐渐增加和PLGA含量的降低. 图2(B)显示了PLGA水凝胶经不同浓度SBF浸泡后的XRD图谱.
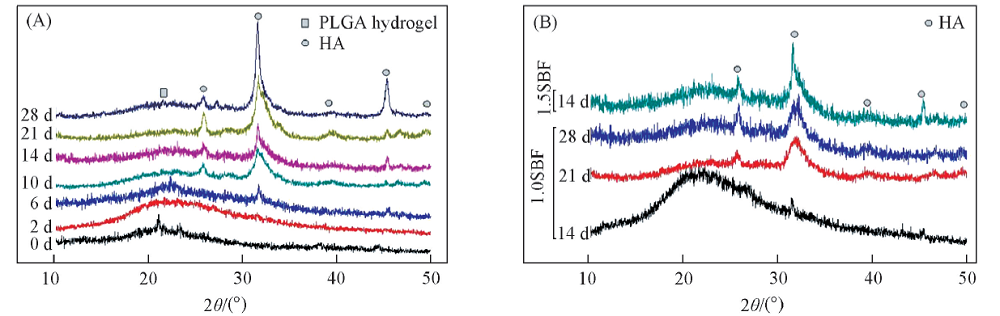 | Fig.2 XRD patterns of PLGA hydrogels after soaking in 1.5SBF for different time(A) and comparison of XRD patterns of PLGA hydrogels after soaking in 1.0SBF and 1.5SBF(B) |
在1.0SBF溶液中浸泡28 d, 仅出现了以2θ =26° 和32° 为中心的2个衍射峰, 其衍射峰强度明显弱于经1.5SBF浸泡14 d的对照组. 因此, 增加模拟体液中的离子浓度可明显加速HA晶体的沉积.
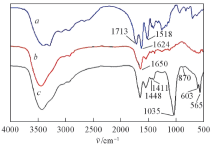
图3为PLGA, PLGA水凝胶以及经1.5SBF浸泡14 d后复合水凝胶的红外光谱图. PLGA在1713, 1624和1518 cm-1处显示特征吸收峰, 分别对应于— COOH、 酰胺Ⅰ 和Ⅱ 以及C=O的伸缩振动[24]. 对于PLGA水凝胶, 1650 cm -1处新峰的出现证实了交联反应发生. PLGA水凝胶经1.5SBF浸泡后, 于1035, 603和565 cm-1处出现了P
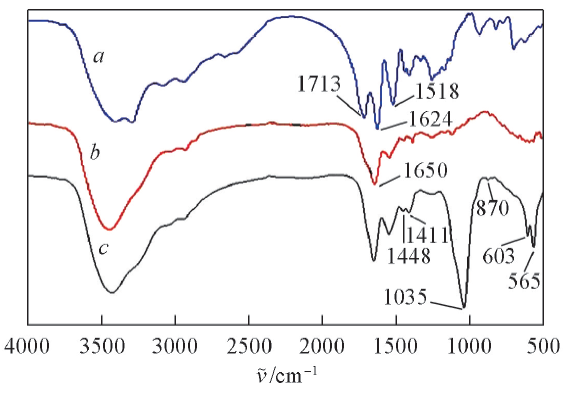 | Fig.3 FTIR spectra of PLGA(a), PLGA hydrogel before(b)and after mineralization(c) |
图4显示了经1.0SBF和1.5SBF溶液浸泡不同时间后PLGA水凝胶的照片. 即使在1.0SBF中浸泡28 d, 水凝胶几乎还是透明的. 然而, 在1.5SBF中浸泡14 d后的PLGA水凝胶透明度就明显变差, 表明HA晶体沉淀. 当浸泡时间增加至28 d时, PLGA水凝胶的透明度进一步降低, 表明更多的HA发生矿化.
'>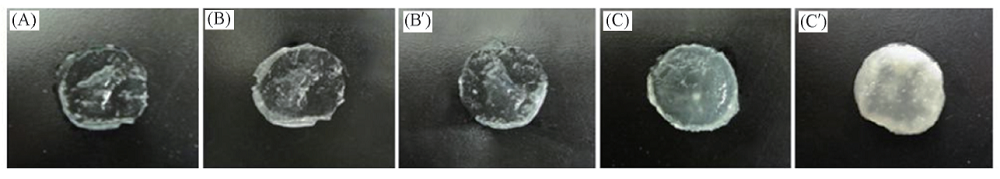 | Fig.4 Photographs of PLGA hydrogels before(A) and after soaking in 1.0SBF(B, B')/1.5SBF(C, C') for 14 d(B, C) and 28 d(B', C') |
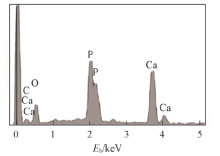
图5显示了经1.0SBF和1.5SBF浸泡不同时间后PLGA水凝胶的表面微观形态. 经1.5SBF浸泡2 d后, 水凝胶表面仍然相对光滑, 只存在少量沉淀物[图5(A')]. 浸泡6 d后, 一些球形颗粒开始出现在水凝胶表面上[图5(B')]. 浸泡10 d后, 颗粒数量和尺寸均增加[ 图5(C')]. 颗粒数随浸泡时间增加进一步增加[图5(D'), (E')]. 浸泡28 d后, 可观察到粒径为35 μ m的球形颗粒几乎覆盖水凝胶外表面[图5(F')]. 而经1.0SBF浸泡后, 在PLGA水凝胶表面上也可观察到球形HA颗粒随浸泡时间延长而逐渐生长[图5(A)(F)], 但与经1.5SBF浸泡样品相比, 其颗粒尺寸和数量都明显变小. 因此, 提高模拟体液中离子浓度可加速HA的沉积. 图5(G)显示了经1.5SBF浸泡14 d后PLGA水凝胶的内部微观结构. 矿化后水凝胶内部仍保持多孔结构, 且水凝胶孔壁覆盖一层致密的球形HA颗粒, 颗粒尺寸为35 μ m. 因此, PLGA水凝胶在模拟体液中的矿化导致在水凝胶内部和表面上形成HA. 图5(H)显示了矿化层的高倍放大形貌. 可见HA球形颗粒的表面显示纳米多孔的微结构[11], 纳米拓扑结构在骨组织工程应用中的重要性已经在成骨细胞分化[34]和细胞黏附等方面[35]得到证实. EDS能谱(图6)显示经浸泡的PLGA水凝胶表面主要元素为C, O, Ca和P, 其中Ca和P只能来自矿化物质. 经1.5SBF浸泡14 d的PLGA水凝胶中Ca/P摩尔比为1.63, 略低于HA的化学计量值1.67. 这可能归因于少量Ca2+位点的阳离子取代, 或P
'> | Fig.5 SEM images of the mineralized surface of PLGA hydrogels after soaking in 1.0SBF and 1.5SBF for different time (A— F) 2, 6, 10, 14, 21 and 28 d in 1.0SBF; (A'— F') 2, 6, 10, 14, 21 and 28 d in 1.5SBF; (G, H) different magnification in 1.5SBF for 14 d. |
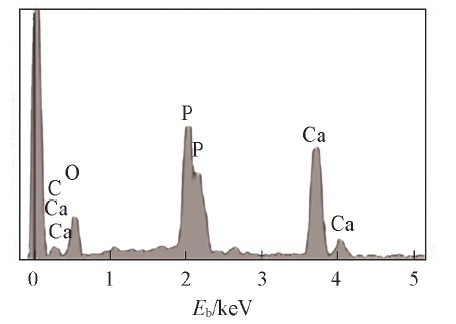 | Fig.6 EDS spectrum of the cross-section of the hydrogel after soaking in 1.5SBF for 14 d |
图7(A)为冷冻干燥的PLGA水凝胶在矿化前后的TGA曲线. 所有样品在25150 ℃和200500 ℃之间均有2个明显的质量损失. 前者归因于干凝胶中残留水分的蒸发, 而后者归因于大分子链的热分解[37]. 在高于500 ℃的温度下复合水凝胶的质量损失不明显, 表明复合水凝胶中的PLGA组分几乎完全分解. 但在500600 ℃时, 剩余质量仍略有下降, 对应于从HA到β -磷酸三钙的热相转变所脱除的结构水[38]. 在1.5SBF溶液中浸泡2 d后, 其TGA曲线与矿化前水凝胶基本一致, 表明PLGA水凝胶基质中几乎没有矿化物. 随着矿化时间的增加, PLGA水凝胶热稳定性大大提高, 这是由于更多的HA沉淀物生成所致[图7(A)]. 由图7(B)可见, 初始分解温度和剩余质量随着矿化时间的延长而增加, 表明PLGA水凝胶的耐热性也随浸泡时间增加而提高. 与经1.0SBF浸泡28 d的PLGA水凝胶相比, 在1.5SBF中浸泡14 d的PLGA水凝胶表现出更高的初始分解温度和残余质量.
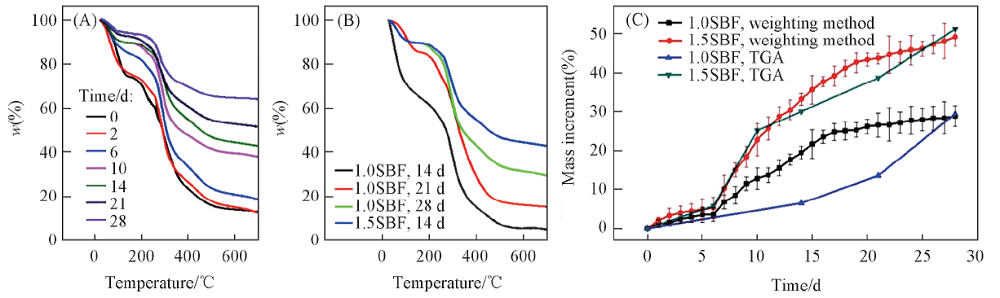 | Fig.7 TGA curves of PLGA hydrogels after mineralization (A) PLGA hydrogels after soaking in 1.5SBF for different time; (B) comparison of TGA curves for PLGA hydrogels after soaking in 1.0SBF and 1.5SBF; (C) percentage of mass increase with immersion time for PLGA hydrogels after soaking in 1.0SBF and 1.5SBF. |
通过称重法和TGA评估矿化后水凝胶质量增加的百分比[图7(C)]. 由于HA的逐渐生长, 在1.0SBF和1.5SBF中矿化的PLGA水凝胶的总质量随着浸泡时间延长而增加. 当浸泡时间从2 d增加到28 d时, 在1.0SBF中矿化的PLGA水凝胶增重率从3.3%增加到29.2%, 而经1.5SBF浸泡的水凝胶的增重率从5.7%增加到49.2%. 在1.5SBF中矿化的PLGA水凝胶的总质量增长得更快, 说明模拟体液中离子浓度的增加可明显加速PLGA水凝胶的矿化. 矿化后水凝胶质量增加也可以通过矿化前后TGA曲线的剩余质量的差值来估算, 在1. 0SBF中浸泡28 d PLGA水凝胶质量增加了29.43%, 而在1.5SBF中增加了51.17%. 2种方法的结果基本相同. 因此, 矿化时间和模拟体液浓度是影响PLGA水凝胶矿化的重要因素.
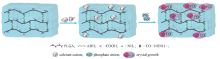
羟基[39]、 羧基、 磷酸根[40]和磺酸根[41]等官能团可作为钙离子结合位点, 从而引发HA晶核的形成[42]. 另一方面, 水凝胶中的氨基却不能使HA成核[42]. HA形成的潜在机制如Scheme 2所示. PLGA中谷氨酸的结构单元富含羧基, 在HA的成核和生长过程中起重要作用[43]. PLGA中的羧基可首先螯合钙离子, 导致PLGA水凝胶表面和内部的Ca2+富集, 然后进一步组装P
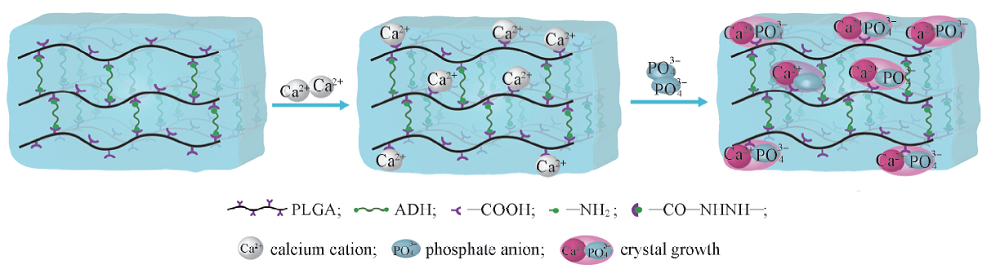 | Scheme 2 Schematic illustration of the HA mineralization induced by PLGA hydrogels Naka K(ed) |
使用旋转流变仪在角频率扫描模式下测试矿化前后PLGA水凝胶的机械性能. 所有样品储能模量(G')在测量频率范围内均远大于损耗模量(G″), 表明PLGA水凝胶形成稳定的交联网络. 矿化前PLGA水凝胶的G'为5.1 kPa, 矿化后水凝胶的模量大大增加. 在1.5SBF中浸泡28 d后, 水凝胶的G'可达45.2 kPa, 其数值比矿化前增加约8.9倍[图8(A)], 这主要归因于PLGA水凝胶中丰富的— COOH促进了Ca2+螯合和HA的沉积, 使水凝胶力学强度提高[45, 46]. Heinemann等[47]也报道了HA矿化对胶原支架的增强效果. Jiao等[45]也报道了HA的引入改善了原位交联的聚(乙二醇)马来柠檬酸酯基水凝胶的力学强度. 动态黏度(|η * |)也随浸泡时间延长明显提高[图8(B)], 这归因于HA的引入限制了大分子链间的相对运动.
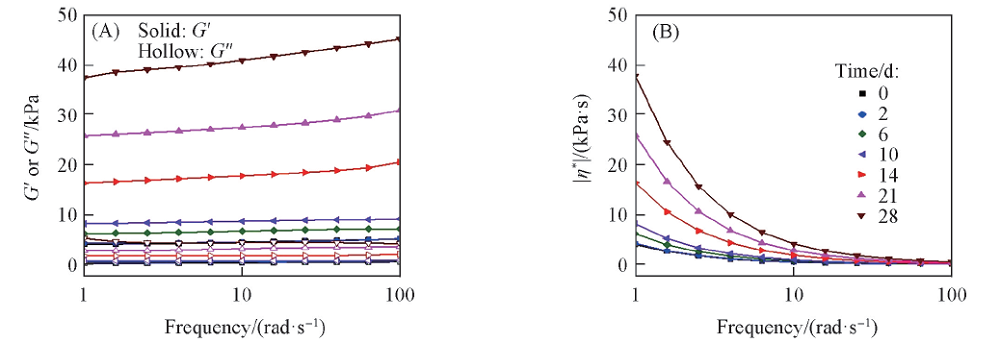 | Fig.8 Rheological properties of PLGA hydrogels after mineralization (A) Storage modulus(G') or loss modulus(G″); (B) complex viscosity |η * | of PLGA hydrogels after soaking in 1.5SBF for different time. |
采用CLSM和SEM研究矿化前后PLGA水凝胶能否为脂肪干细胞(ASC)的培养提供适宜的环境. 矿化前PLGA水凝胶显示出连续多孔结构, 孔径为150200 μ m[图9(A)]. 球形细胞在水凝胶表面上充分增殖并迁移至PLGA水凝胶内部的孔洞中[图9(C)]. 对于在1.5SBF中矿化14 d的PLGA水凝胶, PLGA水凝胶的表面均匀覆盖一层HA, 矿化水凝胶仍保持多孔结构[图9(B)]. 将ASCs在水凝胶中培养3 d后, 可观察到细胞聚集并分泌大量的细胞外基质[48][图9(D)]. 因此, PLGA水凝胶的矿化可促进细胞迁移与生长[49].
 | Fig.9 SEM images of PLGA hydrogels before(A) and after soaking in 1.5SBF for 14 d(B) and the ASCs proliferated on PLGA hydrogel(C) and mineralized hydrogel for 3 d(D) |
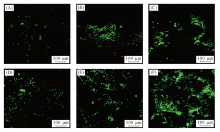
将ASCs用FDA/PI进行死/活细胞染色, 利用CLSM观察不同时间点的细胞-水凝胶复合物. 对于PLGA水凝胶和矿化水凝胶, 球形细胞在7 d内沿着孔壁生长[图10(A), (B), (D), (E)].
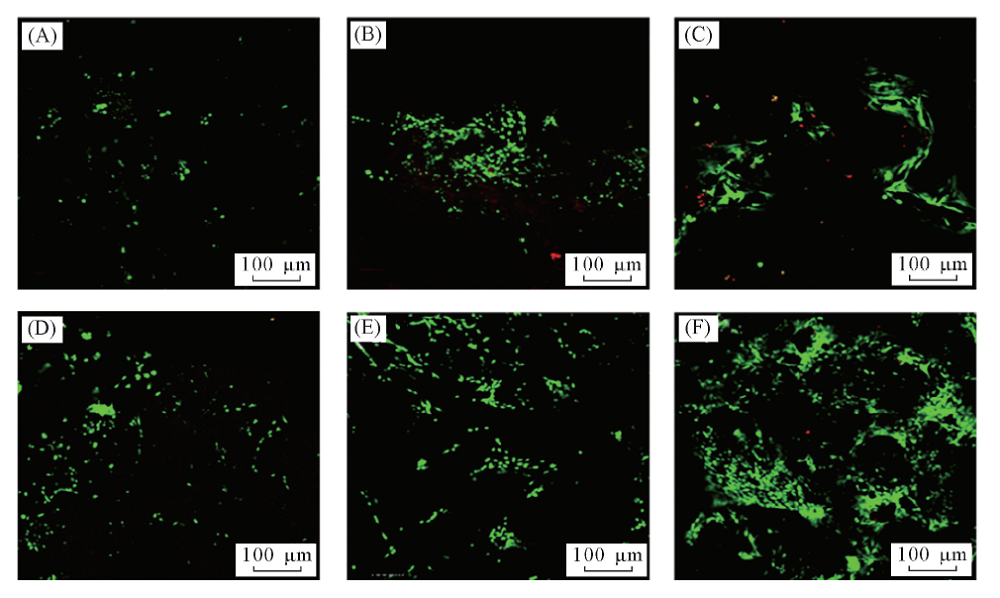 | Fig.10 CLSM images of the ASCs-seeded PLGA hydrogels(A— C) and the ASCs-seeded mineralized hydrogel(D— F) at 1 d(A, D), 7 d(B, C) and 14 d(C, F) The living cells were stained with FDA(green) and the dead cell nuclei were stained with PI(red). |
培养14 d后, 大多数孔洞充满细胞簇, 细胞密度随着培养时间延长而增加[图10(C, F)]. 大多数细胞在1, 7和14 d后仍存活, 证实在矿化前后PLGA水凝胶对细胞均没有明显的毒副作用. 前期的工作[50, 51]也证实, PLGA是一种细胞相容性良好的生物材料, 可促进ASCs的黏附与增殖. 研究[10]表明, 材料的表面特性, 特别是化学成分和形态可影响细胞黏附. 矿化后HA层的引入改变了PLGA水凝胶的化学组成以及表面微观结构, 导致细胞黏附和存活的进一步改善. Chang等[52]也报道了HA的掺入促进了细胞的生长, 并改善了几丁质水凝胶的生物相容性.
以ADH为交联剂, EDC为活化剂, 制备了PLGA水凝胶, 研究了PLGA水凝胶在模拟体液中的生物矿化情况. 结果表明, 具有丰富羧基的PLGA水凝胶可有效诱导HA的形成. 模拟体液浓度和矿化时间的增加显著影响PLGA水凝胶的矿化过程和加速HA的沉积, 且矿化后水凝胶的力学强度明显提高. 矿化前后PLGA水凝胶均具有良好的细胞相容性, 并且用于ASCs的培养. 与PLGA水凝胶相比, 矿化PLGA水凝胶表现出更优的细胞黏附性和细胞相容性, 在骨组织工程领域具有潜在的应用价值.
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|
| [20] |
|
| [21] |
|
| [22] |
|
| [23] |
|
| [24] |
|
| [25] |
|
| [26] |
|
| [27] |
|
| [28] |
|
| [29] |
|
| [30] |
|
| [31] |
|
| [32] |
|
| [33] |
|
| [34] |
|
| [35] |
|
| [36] |
|
| [37] |
|
| [38] |
|
| [39] |
|
| [40] |
|
| [41] |
|
| [42] |
|
| [43] |
|
| [44] |
|
| [45] |
|
| [46] |
|
| [47] |
|
| [48] |
|
| [49] |
|
| [50] |
|
| [51] |
|
| [52] |
|