
{kind=link}
{kind=link}
{kind=link}
TBAC/TBHP体系下6-氨基甲酰基取代的啡啶类化合物的合成
[代佳男, 杨子祺, 魏忠林, 曹军刚, 梁大鹏, 段海峰 , 林英杰]
, 林英杰]
, 林英杰]
|
|


联系人简介: 段海峰, 男, 博士, 教授, 主要从事有机合成方法学方面的研究. E-mail: duanhf@jlu.edu.cn;林英杰, 男, 博士, 教授, 博士生导师, 主要从事有机合成方法学方面的研究. E-mail: linyj@jlu.edu.cn
采用2-异氰基联苯与N,N-二甲基甲酰胺(DMF)作为反应原料, 在四丁基氯化铵(TBAC)、 磷酸氢二钾(K2HPO4)和叔丁基过氧化氢(TBHP)组成的催化氧化体系作用下, 通过一步构建2个C—C键, 以较高的产率合成了一系列6-氨基甲酰基取代的啡啶类化合物(产率高达86%). 同时利用该方法研究了一系列含有推电子和吸电子取代基的2-异氰基联苯衍生物的普适性, 为具有药物活性的该类分子的合成提供了一种新的方法.
In the presence of simple K2HPO4, under catalytic oxidation of tetrabutylammonium chloride(TBAC) and tert-butyl hydroperoxide(TBHP), the couple reaction of 2-isocyanobiphenyl derivatives with N,N-dimethylformamide(DMF) was realized, and 6-aminoformyl-substituted phenanthridine compounds were obtained by formation of two C—C bonds in one pot reaction. A series of 2-isocyanobiphenyl derivatives containg electron-donating and withdrawing substituents was investigated. Corresponding 6-aminoformyl substituted phenanthridines were constructed in moderate to high yields(up to 86% yield), which would provide promising candidates for chemical biology and drug discover.
啡啶核结构存在于许多天然生物碱和重要的药物中[1, 2, 3, 4, 5], 具有该片段结构的化合物通常表现出抗癌性、 抗菌性、 抗白血病性和具有细胞毒性等多种生物活性[6, 7, 8, 9, 10, 11, 12, 13]. 因此, 啡啶类化合物的合成吸引了众多合成化学家的研究兴趣, 相应的构建啡啶衍生物的合成方法相继被报道[14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32]. 另一方面, 氨基甲酰基片段作为一种重要的结构单元也普遍存在于药物、 农药、 天然产物和生物高分子中[33, 34, 35, 36, 37, 38, 39, 40, 41]. 在众多的构建氨基甲酰基的方法中, 最为常用的方法是通过羧酸或羧酸衍生物的酰化反应法[42, 43, 44, 45] . 但这种方法反应效率低、 反应条件苛刻、 反应底物官能团的容忍性不好. 因此, 发展简捷、 高效的构建啡啶环和氨基甲酰基结构的方法尤为迫切[46]. 在前期工作基础上, 我们采用2-异氰基联苯与N, N-二甲基甲酰胺(DMF)为原料, 经催化氧化偶联反应, 一步构建了2个C— C键, 进而完成了系列6-氨基甲酰基取代的啡啶类化合物的合成, 为具有生物和药物活性的6-氨基甲酰基取代的啡啶类化合物设计合成及筛选提供了可能.
实验中涉及到的反应溶剂均经KOH, Na或CaH2纯化, 其余所用试剂均为分析纯.
Varian Mercury 300 BB-300 MHz核磁共振仪; Bruker NMR Spectrometer-400 MHz核磁共振仪; Bruker Agilent 1290 MicroOTOF-QⅡ 型液相色谱-质谱联用仪; 北京泰克X-6型显微熔点测定仪.
1.2.1 2-异氰基联苯类化合物的合成 分别向50 mL三颈瓶中加入苯硼酸类化合物(9.60 mmol)、 邻溴苯胺类化合物(8.00 mmol)和碳酸钾溶液(2 mol/L, 18 mL), 在氩气保护下室温搅拌至溶解并加入乙二醇二甲醚(16 mL), 室温搅拌30 min后快速加入PdCl2(PPh3)2(112.30 mg, 0.16 mmol), 将体系升温到80 ℃后搅拌16 h. 然后降至室温, 以乙酸乙酯(15 mL× 3)萃取, 合并有机相. 有机相分别用水(15 mL× 3)及饱和食盐水(10 mL)洗涤, 用Na2SO4 干燥. 有机相经减压浓缩, 粗产物经柱层析(乙酸乙酯/石油醚体积比1: 30)可得邻氨基联苯化合物.
取等摩尔的甲酸和乙酸酐(14.40 mmol)于干燥的50 mL圆底烧瓶中, 在55 ℃下搅拌2 h, 将反应混合物冷却至室温. 取上述所得邻氨基联苯化合物(6.00 mmol)溶解于12 mL 的干燥四氢呋喃中, 在0 ℃先将该溶液缓慢滴加到圆底烧瓶中, 然后升温至室温反应2 h. 用饱和NaHCO3溶液(20 mL)猝灭反应. 反应混合物以乙酸乙酯(15 mL× 3)萃取, 合并有机相, 分别用水(15 mL× 3)和饱和食盐水(10 mL)洗涤, 用Na2SO4 干燥. 有机相经减压浓缩, 得到酰胺粗产物.
将2.5 mL(18.00 mmol)三乙胺和5 mL干燥的四氢呋喃加入到50 mL 干燥的圆底烧瓶中, 在0 ℃下将上述酰胺粗产物(3.00 mmol)加入到体系中搅拌至其溶解. 保持温度不变, 在2 h内将POCl3(4.5 mmol)缓慢加入到体系中, 于0 ℃下反应2 h. 然后用饱和Na2CO3溶液(20 mL) 猝灭反应. 反应混合物以乙酸乙酯(15 mL× 3)萃取, 合并有机相, 分别用水(15 mL× 3)和饱和食盐水(10 mL)洗涤, 用Na2SO4 干燥. 有机相经减压浓缩, 粗产物经柱层析(乙酸乙酯/石油醚体积比1: 50), 得到异腈类化合物1a1o.
1.2.2 6-氨基甲酰基啡啶类化合物(3a3o) 的合成 分别将四丁基氯化铵(TBAC, 0.5 mmol)、 磷酸氢二钾(0.5 mmol)和2-异氰基联苯类化合物1(0.5 mmol)加入到干燥的DMF(5 mL)中, 然后向体系中加入叔丁基过氧化氢(TBHP, 3.0 mmol), 将体系温度升至90 ℃后继续搅拌6 h. 冷却至室温, 加入适量水稀释该体系, 混合物用乙酸乙酯(20 mL× 3)萃取, 合并有机相. 有机相再分别用水(20 mL× 3)和饱和食盐水(20 mL)洗涤, 用Na2SO4 干燥. 有机相经减压浓缩得粗产物, 经柱层析(乙酸乙酯/石油醚体积比1: 31: 1)得到产物3a3o.
合成化合物的理化性质及核磁共振数据分别列于表1和表2. 其原始谱图见本文支持信息.
| Table 1 Appearance, yields and HRMS data of compounds 3a— 3o |
| Table 2 1H NMR and 13C NMR data of compounds 3a— 3o |
以2-异氰基联苯和DMF作为起始底物, 四丁基碘化铵(TBAI)为催化剂, 以苄基氯化铵盐(DBU)作为碱, 叔丁基过氧化氢(TBHP)作为氧化剂, 其反应式如Scheme 1所示.
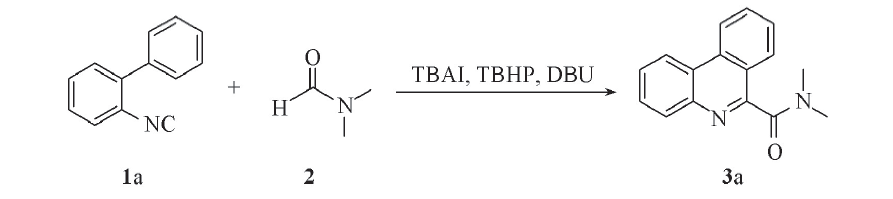 | Scheme 1 Synthesis of compound 3a with TBAI as catalyst, DBU as base and TBHP as oxidant |
考察了参与反应各组分比例对反应的影响, 如表3所示. 1, 1, 2-三氯乙烷(1, 1, 2-TCA)作为溶剂时, 均未发现产物3a(表3中Entries 1 and 2). 当以DMF作为溶剂时, 反应产物的收率最高可达28%(表3中Entry 3 ). 而当反应时间增加到48 h时, 反应产率下降到17%; 反应在624 h时, 产物的收率几乎没有明显变化, 但是反应时间超过24 h后, 产物中可能存在副反应, 从而使反应产率下降(表3中Entries 46). 因此, 选择DMF作为溶剂, 反应时间为6 h进行下一步优化.
| Table 3 Effect of the proportion of each component involved in the reaction and reaction time on yield a |
考察引发剂类别的影响, 实验结果见表4. 当使用四丁基氯化铵(TBAC)作为引发剂时, 产物3a的产率最高达到64%. 因此, 四丁基氯化铵为该反应的最优引发剂.
| Table 4 Effect of different initiators on yield of compound 3aa |
考察不同的氧化剂对该反应的影响, 实验结果见表5 . 当使用叔丁基过氧化氢(TBHP)作为氧化剂时, 产物收率最高达到64%(表5中Entry 1). 而使用过硫酸钾(KPS)或过氧化苯甲酸特丁酯(TBPB)时, 产率也能达到40%或58%(表5中Entries 3和6). 而二叔丁基过氧化物(DTBP)和过氧化二异丙苯(DCP)则不能很好地促进该反应的发生; 过氧化苯甲酰(BPO)几乎不参与反应(表5中Entries 2, 4和5). 不同规格的叔丁基过氧化氢对反应的影响不很明显(表5中Entries 7和8).
| Table 5 Effect of oxidant categories and specifications on yield of compound 3aa |
在最佳氧化剂叔丁基过氧化氢和最佳引发剂四丁基氯化铵作用下, 考察碱种类对反应的影响, 实验结果见表6. 当体系中不加入碱时, 产率仅为9%(表6中Entry 1), 弱碱三乙胺(TEA)未使得反应达到理想效果(表6中Entry 3). 当选用醋酸钠、 磷酸氢二钾和碳酸钾作为碱时, 所得实验结果令人惊喜, 其中磷酸氢二钾给出了最优的产率75%(表6中Entries 46). 当反应在空气中进行时, 反应的产率基本没有变化(表6中Entry 7).
| Table 6 Effect of different catalysts and reaction environment on yield of compound 3a |
通过探索反应时间、 反应组分、 引发剂类别、 氧化剂类别、 氧化剂规格、 催化剂碱类别和不同反应溶剂等因素, 得到了反应的最优条件: 即化合物1a (0.50 mmol), 磷酸氢二钾(0.50 mmol), 四丁基氯化铵(0.50 mmol), 叔丁基过氧化氢(70% 水溶液 3.00 mmol), DMF(5 mL), 在90 ℃下反应6 h.
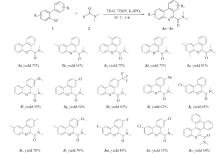
在最佳反应条件下, 探索了该合成方法的普适性. 为此, 合成了15个不同取代的异腈化合物1a1o用于该反应的研究, 实验结果如Scheme 2所示. 可以看出, 该合成法具有良好的底物普适性, 不论吸电子基团(F, Cl, CF3)还是给电子基团(Me和MeO)的2-异氰基联苯类化合物, 均可以中等到优异的收率得到相应的产物. 例如, 当R1为给电子取代基(甲基Me)时或苯环上有多个给电子基(如2个甲基)时, 对应产物3b和3e的产率分别可达61%和81%. 当R1为吸电子取代基团(如Cl)时, 对应产物3j的产率为65%; 当考察苯环上的另一个取代基R2时, 发现一个有趣的现象, 当R2为给电子取代基(Me和OMe)时, 对应产物的产率(3c: 69%, 3d: 74%, 3f: 59%)要高于当R2为吸电子取代基Cl和CF3时所对应产物的产率(3g: 56%, 3h: 65%, 3i: 62%). 这些结果可能归因于, 当 R2为给电子取代基时, 相应苯环的电子云密度会增加, 使得自由基中间体对芳环进行加成反应更加顺利. 考察了化合物1k, 1l, 1m和1n的相应产物都能顺利通过该方法合成得到(3k: 78%, 3l: 59%, 3m: 86%, 3n: 53%). 其中, 当R1和R2同时为F取代基时, 化合物3m产率可达86%. 当R2为稠环时, 对应产物3o分离收率为60%.
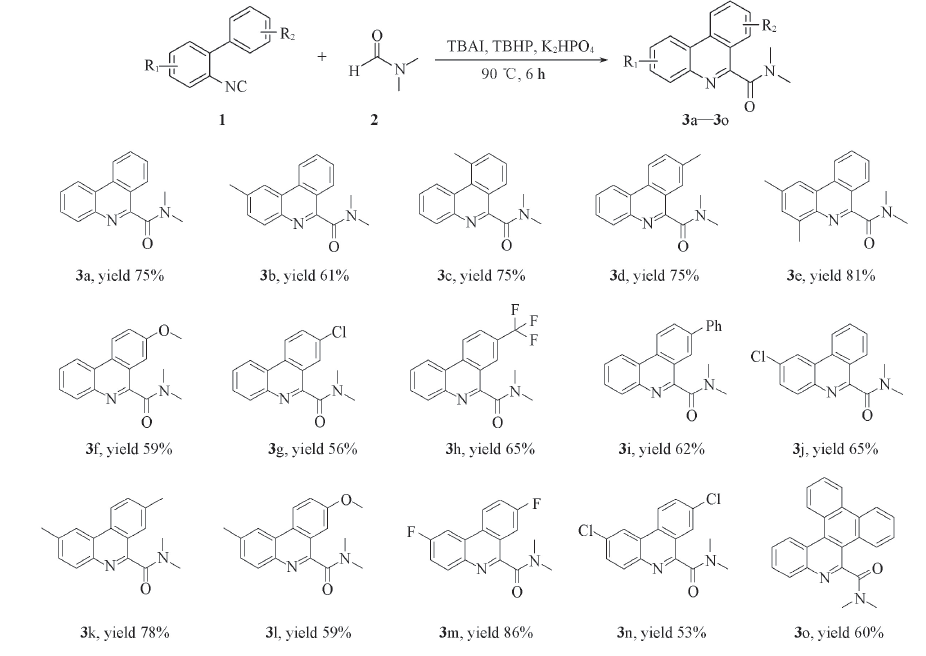 | Scheme 2 Substrate scope of 2-isocyanobiphenyls Reaction conditions: 1a(0.50 mmol), K2HPO4(0.50 mmol), TBAI(0.50 mmol), TBHP(70% aqueous solution 3.00 mmol), DMF(5 mL), 90 ℃ 6 h. Isolated yield. |
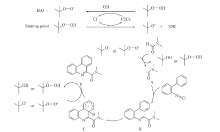
由此, 我们提出了该反应的可能反应机理, 见Scheme 3. TBHP经过TBAC的作用产生叔丁氧基自由基(t-BuO· )或叔丁基过氧自由基(t-BuOO· ), DMF中的C— H键在这些自由基的作用下, 生成酰胺基自由基A. 异腈在A的作用下, 生成中间体B. 中间体B发生分子内自由基环化, 生成中间体C, 经过芳构化生成最终产物.
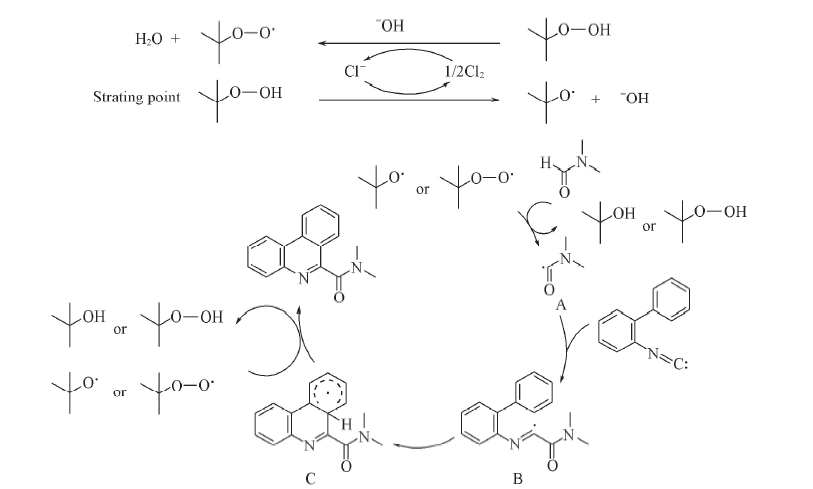 | Scheme 3 Proposed mechanism of reaction |
我们合成了一系列6-甲酰胺基啡啶类化合物. 在四丁基氯化铵/叔丁基过氧化氢/磷酸氢二钾的催化氧化下, 一系列2-异腈基联苯类化合物和DMF进行氧化偶联反应, 同时构建2个C— C键, 以中等到优异的产率合成得到6-甲酰胺基啡啶类化合物(产率高达86%). 该反应提高了操作的简捷性, 同时提升了官能团容忍性, 还避免使用过渡金属, 是一种原子经济性的反应.
支持信息见http://www.cjcu.jlu.edu.cn/CN/10.7503/cjcu20180663.
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|
| [20] |
|
| [21] |
|
| [22] |
|
| [23] |
|
| [24] |
|
| [25] |
|
| [26] |
|
| [27] |
|
| [28] |
|
| [29] |
|
| [30] |
|
| [31] |
|
| [32] |
|
| [33] |
|
| [34] |
|
| [35] |
|
| [36] |
|
| [37] |
|
| [38] |
|
| [39] |
|
| [40] |
|
| [41] |
|
| [42] |
|
| [43] |
|
| [44] |
|
| [45] |
|
| [46] |
|