


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
织构气体扩散层表面氢氧燃料电池阴极超低Pt载量催化层的磁控溅射法制备
[刘家明, 傅凯林, 张泽, 郭伟 , 潘牧]
, 潘牧]
, 潘牧]
|
|


联系人简介: 郭伟, 男, 博士, 副研究员, 主要从事质子交换膜燃料电池研究. E-mail: guowei2016@whut.edu.cn
采用磁控溅射技术在具有织构结构的气体扩散层(GDL)表面制备了可应用于氢氧质子交换膜燃料电池的超低Pt载量阴极催化层, 并通过SEM、 轮廓仪和XRD等测试方法表征了GDL及其载Pt后的形貌和物相, 利用XPS分析溅射Pt的化学价态, 使用电池测试台表征其电池性能. 测试结果表明, 磁控溅射法在GDL表面沉积的Pt催化层载量可控且分布均匀; 与商业GDL对比, Pt在织构GDL表面具有更大的可附着面积. 电池性能测试结果显示, 当Pt载量为0.04 mg/cm2时, 以织构GDL作基材的样品质量比功率高达26.25 kW/g Pt, 远大于商业GDL作基材时的17.76 kW/g Pt, 也大于同等Pt载量下商业Pt/C催化剂的24.00 kW/g Pt.
Ultra-low loading of Pt was deposited on the textured gas diffusion layer(GDL) by magnetron sputtering method to prepare cathodic catalyst layer for hydrogen-oxygen proton exchange membrane fuel cells(PEMFCs). The characterization of GDL before and after Pt-loading was carried out using scanning electron microscope(SEM), profilometer and X-ray diffractometer(XRD). The chemical states of sputter deposited Pt were investigated by X-ray photoelectron spectroscopy(XPS). The performance of membrane electrode asembly(MEA) was tested by fuel cell station. The results showed that the Pt deposited on the GDL was controllable and evenly distributed. The surface area of Pt deposited on the textured GDL was larger than that deposited on the commercial GDL. The fuel cell performance indicated that the Pt deposited on the textured GDL with the loading of 0.04 mg/cm2 exhibited the maximum mass specific power of 26.25 kW/g Pt, which was higher than that of the Pt deposited on the commercial GDL(17.76 kW/g Pt). In addition, the maximum mass specific power of commercial Pt/C was 24.00 kW/g Pt.
质子交换膜燃料电池(PEMFC)因具有广阔的应用前景而备受关注[1]. 其中, 氢氧PEMFC由于具有更高的能量密度, 在航空航天和潜艇潜航等特殊运行环境下拥有巨大的需求[2]. 膜电极(MEA)是氢氧PEMFC的核心部件, 一般采用贵金属Pt作为催化剂. 氢氧膜电极通常采用气体扩散电极(GDE)工艺制备, 但这种工艺制备的阴极Pt载量较高(一般大于1.0 mg/cm2, 多数在2.0~4.0 mg/cm2之间[3]), 因此降低Pt载量依然是氢氧膜电极开发的主要研究方向[4, 5].
为了达到降低Pt载量的目标, 许多科研工作者致力于开发新型工艺来制备氢氧膜电极, 如通过磁控溅射技术或喷涂技术改良GDE工艺[6, 7]或者采用三合一(CCM)工艺[8, 9], 其中磁控溅射沉积技术具有制备工艺简单、 灵活性高、 制备的催化剂材料形貌和结构可控、 溅射基材种类多等优势, 是实现超低Pt载量膜电极最有潜力的制备技术之一[10]. 以气体扩散层(GDL)作为溅射基材, 直接在GDL表面沉积Pt的制备过程非常简单, 更容易实现产业化, 因此, 已有大量关于在GDL表面溅射沉积Pt制备低Pt载量膜电极的研究报道[11, 12, 13, 14, 15]. 1997年, Shinichi等[11]首次用磁控溅射技术在GDL表面沉积0.04 mg/cm2超低载量Pt, 但是由于电池内阻太高, 导致电池性能较差. 后来, Brault等[12]用低压超高频感应等离子溅射系统在GDL表面溅射沉积0.08 mg/cm2的Pt并用于电池阴极侧, 在氢氧测试条件下, 电压0.6 V时的电流密度为0.67 A/cm2, 在高电流密度区域, 电压下降非常迅速. 在GDL表面共溅射沉积Pt和C虽然可将Pt载量降低至0.01 mg/cm2, 但是并没有提升电池性能[13]. Sung等[14]用高压溅射技术在GDL表面溅射沉积0.02 mg/cm2的Pt并分别用于电池的阳极和阴极侧, 在氢氧测试条件、 电压为0.6 V时电流密度为0.65 A/cm2, 最大功率密度为0.42 W/cm2. 上述研究报道均采用商业GDL作为溅射基材, 虽然可以实现超低Pt载量, 但电池性能并不理想.
商业化GDL通常由基底和微孔层(MPL)组成, 基底通常采用碳布或碳纸材料, MPL主要为碳粉与PTFE的混合物. 目前, 商业GDL表面微孔层整体较为平整, 作为溅射基底材料能为Pt粒子附着提供的面积较小, 使Pt粒子更容易堆积, Pt的利用率受限. 而在相同Pt载量下, 织构GDL表面呈高低起伏结构, 能为Pt粒子附着提供更大的面积, 使能参与催化反应的Pt的数量更多, 大大提高Pt的利用率, 从而提高电池性能.
本文以采用丝网印刷涂布技术制备的织构GDL为溅射基材, 利用磁控溅射技术制备了超低Pt载量阴极催化层并应用于氢氧燃料电池, 研究了溅射基材和溅射Pt载量对电池性能的影响, 并与商业Pt/C催化剂的商业CCM制备工艺进行了对比.
Pt靶(纯度99.99%, 贵研铂业股份有限公司); Nafion溶液(质量分数0.5%, 美国Dupont公司); 商业Pt/C(Pt质量分数29.7%, 美国Johnson Matthey公司); MPL浆料(质量分数: C 80%, PTFE 20%, 武汉理工新能源有限公司); 商业GDL(Sigracet 25 BC, 美国SGL公司); 东丽碳纸(Tory, 日本东丽公司).
全自动磁控镀膜系统(TRP-450, 中国科学院沈阳科学仪器有限公司); 全谱直读等离子体发射光谱仪(Prodigy 7, 美国利曼-徕伯斯公司); 场发射扫描电子显微镜(Zeiss Ultra Plus, 德国蔡司公司); 转靶X射线衍射仪(D8 Advance, 德国布鲁克AXS公司); X射线光电子能谱仪(ESCALAB 250Xi, 美国赛默飞世尔科技有限公司); 光学轮廓仪(NPFLEX 3D, 美国布鲁克公司); 群翌测试系统(HTS-125, 中国台湾群益公司, 测试温度为80 ℃, 阳极和阴极分别通入氢气和氧气, 气体流量分别为200和80 mL/s, 过量系数分别为1.5和2.0, 背压均为150 kPa, 相对加湿度均为100%).
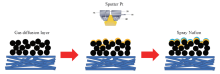
利用全自动磁控镀膜系统制备载铂GDL. 选用尺寸为Φ 60 mm的Pt靶, 靶基距为90 mm, 真空度为3× 10-4 Pa, 工作气压(Ar)为2 Pa, 直流电源溅射功率为20 W, 溅射时间分别为200和1200 s, 基体材料分别为商业GDL和织构GDL. 其中织构GDL是通过丝网印刷的方式将MPL浆料涂覆在东丽碳纸表面, 然后进行烘干处理而获得, 从而使其MPL表面具有织构结构. 按图1所示的工艺流程制备样品, 并分别标记为C-GDL-200 s(商业GDL, 溅射200 s)、 C-GDL-1200 s(商业GDL, 溅射1200 s)、 T-GDL-200 s(织构GDL, 溅射200 s)和T-GDL-1200 s(织构GDL, 溅射1200 s).
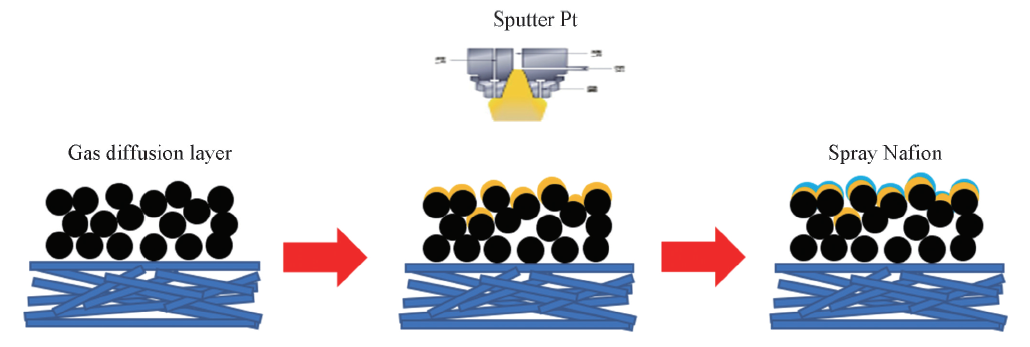 | Fig.1 Flow chart of sputter Pt on the GDL surface |
为了测定在GDL表面溅射不同时间对应的Pt载量, 对一系列载铂GDL样品截取面积为2 cm× 2 cm的小块, 并分别进行全谱直读等离子体发射光谱(ICP-OES)测试, 结果表明, 溅射200和1200 s对应的Pt载量分别为0.04和0.24 mg/cm2.
选用Nafion 211膜(美国Dupont公司)作为质子交换膜, 膜电极活性面积为5 cm× 5 cm. 阳极侧选用商业Pt/C作为催化剂, Pt载量为0.4 mg/cm2, 通过热压转印到膜的一侧表面, 热压温度为150 ℃, 压力1.5 MPa, 时间150 s; 以商业GDL作为气体扩散层; 阴极侧选用上述4种载铂GDL样品, 喷涂质量分数为0.5%的Nafion溶液后置于膜的另一面, 制备成MEA. 另外, 为了与商业Pt/C催化剂对比, 阴极用Pt载量分别为0.04和0.24 mg/cm2的商业Pt/C催化剂制备2个标准MEA, 分别标记为Pt/C-0.04和Pt/C-0.24. 将制备的MEA组装成单电池, 使用群翌测试系统测试电池性能.
为了研究不同GDL表面溅射沉积Pt前后的形貌变化, 对样品进行了SEM测试, 结果如图2所示. 商业GDL在低倍数下看表面较平整[图2(A)], 在高倍数下可见碳颗粒尺寸约为30 nm[图2(B)]; 由溅射200 s时商业GDL的SEM照片可见, Pt覆盖在碳颗粒表面, 使颗粒尺寸略微增大至50 nm左右; 溅射1200 s时的商业GDL表面Pt载量增多, 平均颗粒尺寸增大为90 nm[图2(D)]; 织构GDL在低倍数下呈高低起伏的网格状, 方形格子边长约为175 μ m, 沟壑宽度约为75 μ m[图2(E)], 在高倍数下碳颗粒尺寸约为30 nm[图2(F)]; 溅射200 s时的织构GDL颗粒尺寸约为50 nm[图2(G)]; 溅射1200 s时的织构GDL颗粒尺寸约为90 nm[图2(H)]. 可见, 低倍数下织构GDL与商业GDL表面形貌差异较大, 高倍数下溅射Pt前后的织构GDL表面颗粒尺寸与商业GDL基本相同.
 | Fig.2 SEM images before and after sputtering Pt on GDL surface (A), (B) C-GDL; (C) C-GDL-200 s; (D) C-GDL-1200 s; (E), (F) T-GDL; (G) T-GDL-200 s; (H)T-GDL-1200 s. |
为了进一步量化粗糙度, 采用多区域轮廓仪对2种GDL进行测试, 商业与织构GDL表面的3维图谱如图3所示. 结果显示, 商业与织构GDL的Ra值(算术平均粗糙度)分别为1.83和2.35 μ m. 可见, 织构GDL表面更粗糙, 溅射Pt可附着的表面积更大.
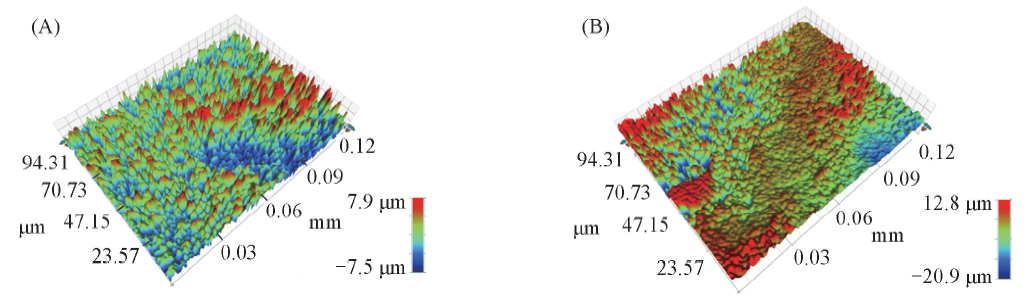 | Fig.3 Profiler images of C-GDL(A) and T-GDL(B) surfaces |
图4给出了不同GDL表面溅射200 s和1200 s后的断面SEM照片以及相同区域的Pt元素分布图. 从图4(A1, A2, B1, B2)可知: 商业GDL表面较平整, Pt分布在GDL表层, 溅射时间越长, Pt的数量越多. 从图4(C1, C2, D1, D2)可知, 织构GDL表面呈高低起伏状, Pt随着织构轮廓均匀分布在GDL表面, 相比于商业GDL, Pt覆盖的面积更大.
 | Fig.4 SEM images(A1— D1) and Pt element distribution maps(A2— D2) of GDL section (A1, A2) C-GDL-200 s; (B1, B2) C-GDL-1200 s; (C1, C2) T-GDL-200 s; (D1, D2) T-GDL-1200 s. |
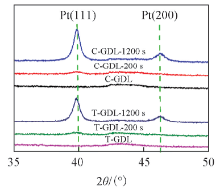
为了进一步验证覆盖在碳颗粒表面的物质为金属Pt, 对溅射前后的GDL样品进行XRD测试, 结果如图5所示. 从XRD谱图中可以看出: 溅射Pt后, 当溅射时间为1200 s时, Pt(111)衍射峰最强, Pt(200)衍射峰较弱, 这与文献报道的结果一致[16]. 而溅射200 s的样品没有明显的Pt衍射峰, 这可能是由于溅射时间非常短, Pt的载量很低导致的.
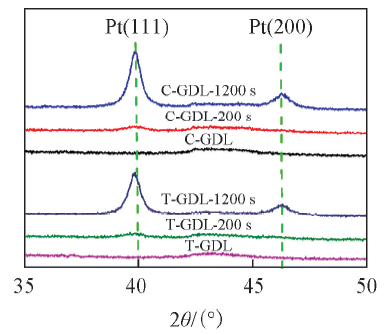 | Fig.5 XRD patterns of Pt sputtered on GDL surface |
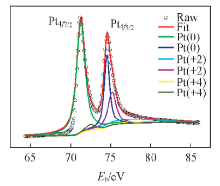
为了表征溅射Pt的化学环境, 对结构GDL表面溅射Pt的样品进行XPS测试, 结果如图6所示. P
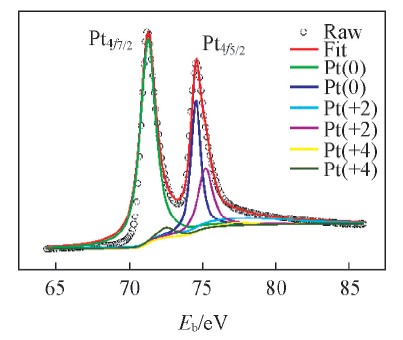 | Fig.6 XPS spectra of Pt sputtered on FGDL surface |
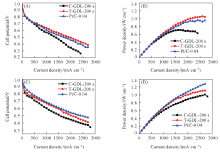
图7示出了商业GDL和织构GDL分别溅射不同Pt载量以及商业Pt/C催化剂的电池性能. 由图7(A)和(B)可知, 当Pt载量为0.04 mg/cm2时, 以商业GDL作为溅射基材的电池在电流密度为1700 mA/cm2时率密度可达0.71 W/cm2, Pt的质量比功率为17.76 kW/g Pt(质量比功率可通过最大功率密度/载量计算得出); 以织构GDL作为溅射基材的电池在电流密度为2400 mA/cm2时功率密度可达1.05 W/cm2, Pt的质量比功率为26.25 kW/g Pt; 商业Pt/C的电池在电流密度为2400 mA/cm2时功率密度达到0.96 W/cm2, Pt的质量比功率为24.00 kW/g Pt, 织构GDL作为溅射基材的电池性能最佳. 这一方面是由于商业GDL表面微孔层较为平整, 而织构GDL表面微孔层呈高低起伏的网格状, 溅射Pt层附着在织构GDL上的面积更大, 一定程度上提高了Pt的覆盖面积, 而且在氢氧条件下, 织构GDL表面的沟壑更有利于水管理; 另一方面是由于在0.04 mg/cm2超低Pt载量的条件下, 溅射Pt厚度约为20 nm, 而商业Pt/C催化层厚度通常为10 μ m左右, 超薄催化层拥有更小的氧传输阻力, 因此, 以织构GDL作为溅射基材制备的超低Pt载量阴极催化层应用于氢氧燃料电池相比以商业GDL作基材和商业Pt/C催化剂具有明显优势.
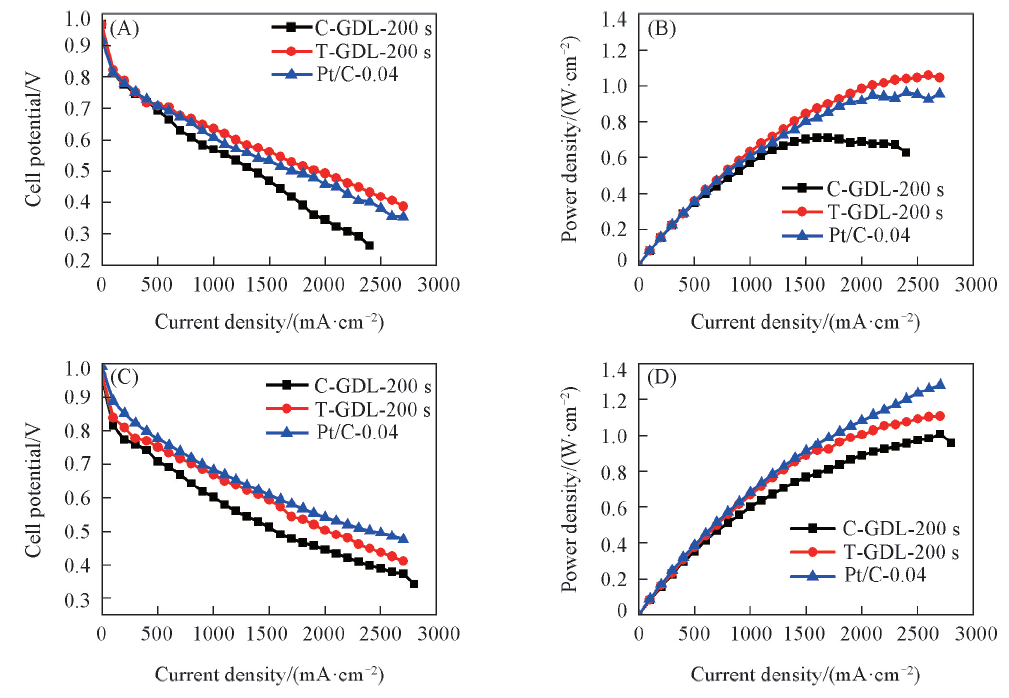 | Fig.7 I-V curves(A, C) and power density curves(B, D) of 0.04 mg/cm2 Pt loading(A, B) and 0.24 mg/cm2 Pt loading(C, D) GDL |
由图7(C)和(D)可知: 当Pt载量增加到0.24 mg/cm2时, 商业GDL作为溅射基材的电池最大功率密度提升到1 W/cm2, Pt的质量比功率为4.16 kW/g Pt; 织构GDL作为溅射基材的电池最大功率密度提升到1.10 W/cm2, Pt的质量比功率为4.58 kW/g Pt; 商业Pt/C催化剂的电池最大功率密度提升到1.26 W/cm2, Pt的质量比功率为5.25 kW/g Pt, 商业Pt/C催化剂的电池性能最佳. 这是因为商业Pt/C上Pt颗粒的尺寸约为3 nm[20], Pt载量越高, 催化层中的Pt颗粒越多, 参与催化反应的Pt表面增大, 性能提升非常明显; 但对于溅射Pt而言, 溅射时间越长, Pt堆积得越厚, Pt载量对应增加, 但只是最外层的Pt参与催化反应, 内层的Pt仅起导电作用, 因此, 溅射Pt堆积得越厚, Pt的质量比功率反而急剧降低.
磁控溅射在GDL表面沉积的Pt载量可控, 织构GDL表面Pt的可附着面积大于商业GDL. 溅射Pt载量为0.04 mg/cm2时, 以织构GDL作基材的电池在电流密度为2400 mA/cm2时功率密度达到1.05 W/cm2, 催化剂的质量比功率为26.25 kW/g Pt, 远大于商业GDL作基材且电流密度为1700 mA/cm2时的功率密度(0.71 W/cm2)和质量比功率(17.76 kW/g Pt), 并大于商业Pt/C催化剂在同载量条件下电流密度为2400 mA/cm2时的功率密度(0.96 W/cm2)和质量比功率(24.00 kW/g Pt).
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|
| [20] |
|