


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
CeTiO x脱硝催化剂的抗硫中毒性能
[朱红太2 , 宋丽云1, 2 , 何洪1, 2  , 尹孟奇
, 尹孟奇2 , 程杰2 , 孙炎明2 , 李世宁2 , 邱文革1, 2 ]
, 尹孟奇|
|


联系人简介: 何 洪, 男, 博士, 教授, 博士生导师, 主要从事低温选择性催化还原脱硝技术方面的研究. E-mail: hehong@bjut.edu.cn
分别以Ce2(C2O4)3和Ce(SO4)2为Ce前驱体, 采用固相球磨法制备了Ce基选择性催化还原(SCR)脱硝催化剂CeTiO x-A[以Ce2(C2O4)3为前驱体]和CeTiO x-B[以Ce(SO4)2为前驱体]. 将2个催化剂分别在体积分数为0.15%的SO2的气氛下反应40和60 h, 得到高硫条件下SCR反应后的CeTiO x催化剂, 分别记为40CeTiO x-A, 60CeTiO x-A, 40CeTiO x-B和60CeTiO x-B. 考察了反应前后催化剂的NH3-SCR反应活性. 采用X射线衍射(XRD)、 X射线荧光光谱(XRF)、 比表面积(BET)测试、 H2程序升温还原(H2-TPR)、 X射线光电子能谱(XPS)、 NH3程序升温脱附(NH3-TPD)及SO2程序升温脱附(SO2-TPD)等技术对样品进行了表征. 结果表明, CeTiO x-A系列催化剂比CeTiO x-B系列催化剂具有更高的NH3-SCR反应活性和更好的抗硫抗水性能. 与CeTiO x-B系列催化剂相比, CeTiO x-A系列催化剂具有更大的比表面积和孔容, 更多的Ce3+和吸附氧(O α)物种有助于NO的吸附和活化; CeTiO x-A系统催化剂还具有更多的Lewis强酸性位数量, 可以吸附更多的NH3分子, 有利于催化剂上NH3-SCR反应的进行, 提高了CeTiO x-A系列催化剂的NO转化率.
, YIN MengqiCeria-based catalysts(CeTiO x-A and CeTiO x-B) were prepared by solid state ball milling method with Ce2(C2O4)3 and Ce(SO4)2 as precursors, respectively. Then, the prepared ceria-based catalysts were treated in reaction atmosphere consisted of 0.15%SO2 for 40 and 60 h, respectively. The obtained catalysts were designated as 40CeTiO x-A, 60CeTiO x-A, 40CeTiO x-B and 60CeTiO x-B. The performances of fresh and used catalysts for selective catalytic reduction(SCR) of NO with NH3 as the reductant were investigated. The catalysts were also characterized using X-ray diffraction(XRD), X-ray fluorescence spectra(XRF), Brunner emmet teller(BET), H2-temperature programmed reduction(H2-TPR), X-ray photoelectron spectra(XPS), NH3-temperature programmed desorption(NH3-TPD) and SO2-temperature programmed desorption(SO2-TPD) techniques. The results indicated that the NH3-SCR activity, SO2 and H2O resistance over the CeTiO x-A catalysts were better than over the CeTiO x-B samples. The reasons for this phenomenon are that CeTiO x-A catalysts possess larger specific surface area and pore volume than CeTiO x-B catalysts. Besides, CeTiO x-A catalysts have plenty of Ce3+ and the surface adsorbed oxygen, which would benefit the adsorption and activation of NO. Furthermore, CeTiO x-A catalysts also have numerous strong Lewis acid sites, which lead to the increasing adsorption of NH3. Therefore, the NH3-SCR reaction rate and the NO conversion of CeTiO x-A catalysts was improved.
近年来, 我国大部分地区雾霾污染频发, 氮氧化物(NOx)是其主要污染源之一. 如何降低NOx的排放是当前备受关注的课题. 选择性催化还原技术(SCR)是目前去除NOx的主要手段. 商业SCR催化剂中最常用的是V2O5-WO3(MoO3)/TiO2系列催化剂, 但它存在一些缺陷: 活性组分V2O5具有较大的毒性, 为废旧催化剂的处理带来了困难. 对于传统的SCR催化剂, 当运行温度较高时, 烟气中的SO2易氧化为SO3, 且催化剂的N2选择性降低, 易生成N2O[1, 2, 3, 4]. 因此, 开发新型非钒基 SCR催化剂, 并将其实际应用至关重要. 目前, 许多金属(Ce, Fe, Ti, Cu, W和Mo等)氧化物SCR催化剂均表现出优异的NH3-SCR反应活性, 得到了广泛研究. 其中, Ce基SCR催化剂因具有优异的氧化还原性能而备受关注[5, 6, 7, 8]. Gao等[9]分别用溶胶-凝胶法、 浸渍法和共沉淀法制备了CeO2-TiO2催化剂, 实验结果表明, 溶胶-凝胶法制备的CeO2-TiO2催化剂具有最高的NO转化率和最好的抗SO2中毒能力, 主要是因为在该法制备的催化剂中CeO2呈无定形态高度分散在TiO2表面. Zhang等[10]发现, 在CeO2中加入H3PO4· 12WO3· xH2O, 可以显著提高催化剂的NH3-SCR反应活性和N2选择性; 在150~550 ℃范围内, 催化剂的NO转化率达到90%以上, 且催化剂的抗H2O和SO2中毒能力也明显提高. Gao等[11]用浸渍法制备了CeO2-ZrO2催化剂, 研究了硫化后CeO2-ZrO2催化剂与新鲜催化剂的SCR脱硝活性的差别, 发现硫化后的CeO2-ZrO2催化剂的NO转化率由未硫化的70%提高至95%, 且表面具有大量的酸性位, 显著提高了催化剂抗碱金属中毒的能力. Zhang等[12]研究了CeO2的硫化温度对其NH3-SCR反应活性的影响, 发现随着硫化温度的升高, NO转化率逐渐降低, CeO2表面生成的类体相硫酸盐和体相硫酸盐是催化剂脱硝活性降低的主要原因. 前文[13]采用有机配体辅助球磨法制备的CeO2/TiO2催化剂具有较高的低温活性和较好的抗硫抗水性能. 为了更加深入地考察该类催化材料的抗硫中毒能力, 本文选择2种Ce前驱体, 采用固相球磨法制备了CeTiOx催化剂, 并在含有高浓度SO2的反应气氛下反应40和60 h, 比较了反应前后催化剂的NH3-SCR反应活性, 并采用X射线衍射(XRD)、 X射线荧光光谱(XRF)、 比表面积测试(BET)、 H2程序升温还原(H2-TPR)、 X光电子能谱(XPS)、 NH3程序升温脱附(NH3-TPD)和SO2程序升温脱附(SO2-TPD)等技术对样品进行了表征.
草酸铈[Ce2(C2O4)3· 9H2O], A.R.级, 国药集团化学试剂有限公司; 硫酸铈[Ce(SO4)2], A.R.级, 上海阿达玛斯试剂有限公司; 二氧化钛(TiO2), 工业级, 重庆新华化工有限公司.
Tensor 27型傅里叶红外光谱(FTIR)仪, 德国Bruker公司; GC-2014C型气相色谱仪, 日本Shimadzu公司; SIGNAL 4000VM型氮氧化物分析仪, 50 kV, 50 mA, 英国Signal Instruments公司; D8 ADVANCE型X射线粉末衍射仪, Cu Kα (λ =0.154 nm), 50 kV, 35 mA, 2θ =10° ~80° , 扫描速率3.5° /min, 德国Bruker公司; Magix PW2403型X射线荧光光谱分析仪, 荷兰PANalytical公司; ASAP 2020型全自动物理吸附仪和Autochem Ⅱ 2920型化学吸附仪, 美国Micromeritics公司; ESCALAB 250 Xi型X射线光电子能谱仪, 美国Thermo Fisher公司; ChemBET Pulsar TPR/TPD型化学吸附仪, 美国Quantachrome公司.
采用固相球磨法制备催化剂. 称取定量的Ce2(C2O4)3· 9H2O和Ce(SO4)2, 分别和一定量的TiO2放入球磨罐中, 加入直径为10 mm的陶瓷球, 陶瓷球与试剂的质量比约为12: 1, 设定球磨机转速为500 r/min, 球磨时间1 h. 将球磨后的粉体置于马弗炉中, 在空气气氛下于250 ℃焙烧2 h, 再于500 ℃焙烧2 h, 即得到CeTiOx-A[以Ce2(C2O4)3为前驱体]和CeTiOx-B[以Ce(SO4)2为前驱体]SCR催化剂粉体. 将得到的粉体压片, 粉碎, 筛取20~40目颗粒备用. 催化剂中CeO2质量分数为30%. 分别将CeTiOx-A和CeTiOx-B催化剂在下列条件下反应40和60 h, 气体组成(体积分数): NO(0.1%)+NH3(0.1%)+SO2(0.15%)+O2(6%), 氮气为平衡气, 气体总流量为500 mL/min, 空速(GHSV)为4000 h-1, 温度为300 ℃, 得到高硫条件下SCR反应后的CeTiOx催化剂, 分别记为40CeTiOx-A, 60CeTiOx-A, 40CeTiOx-B和60CeTiOx-B.
催化剂的NH3-SCR催化反应活性和寿命评价在固定床微型反应器(ϕ =6 nm)中进行, 用石英棉将1 mL催化剂(20~40目)固定在反应器中, 催化剂床层上端装有热电偶, 用于控制和监测反应温度, 将反应器置于带有程序升温装置的管式炉内. 反应气体组成: NO(0.1%)+NH3(0.1%)+O2(6%), He气为平衡气, 在需要时通入SO2(0.0175%)和H2O(6%), 气体总流量为500 mL/min, 空速(GHSV)为3× 104 h-1. 在评价催化剂活性时, 反应后各气体组分(N2, NO, NO2和N2O)的浓度用配有2.4 m光程气体池的傅里叶红外光谱仪和气相色谱仪检测[14]. 在评价催化剂寿命时, SCR反应温度为300 ℃, 反应后各气体组分用氮氧化物分析仪测定.
NO转化率和N2选择性通过下式计算[15]:
式中: [NO]in(%)和[NO]out(%)分别为SCR反应器入口和出口的NO平均浓度; [NO2]out(%)为SCR反应器出口的NO2平均浓度; [N2O]out(%)为SCR反应器出口的N2O平均浓度.
1.4.1 催化剂的比表面积、 孔容、 孔径和孔径分布测试 取200 mg催化剂粉末样品在200 ℃真空条件下预处理2 h, 在-196 ℃下进行N2静态吸附实验, 采用BET法计算催化剂的比表面积, 采用BJH(Barrett-Joyner-Halenda)法计算催化剂的孔径分布.
1.4.2 H2程序升温还原(H2-TPR)实验 取50 mg催化剂样品(20~40目), 在空气气氛下, 于300 ℃预处理30 min, 降温至40 ℃后, 切换为Ar气吹扫30 min, 然后通入10%H2/Ar气, 待仪器基线平稳后, 以10 ℃/min的速率升温进行测试, 采用热导检测器(TCD)在线记录H2浓度随温度变化的曲线, H2消耗量根据氧化铜标样来计算.
1.4.3 催化剂表面元素价态测试 以Al Kα 的X射线为激发光源, 测定催化剂表面物种中Ti2p, S2p, Ce3d和O1s的电子结合能(Eb), 以表面污染碳的C1s电子结合能(284.6 eV)校正电子结合能数据.
1.4.4 NH3程序升温脱附(NH3-TPD)实验 取200 mg催化剂粉末样品, 在He气氛下于450 ℃预处理60 min, 降温至40 ℃后, 切换为5%NH3/He气吸附30 min, 然后切换He气吹扫, 待仪器基线平稳后, 以10 ℃/min的速率升温进行测试, 采用热导检测器检测NH3的脱附信号.
1.4.5 SO2程序升温脱附(SO2-TPD)实验 取100 mg粉末催化剂样品, 在N2气氛下于200 ℃预处理60 min, 降温至40 ℃后, 切换为0.5%SO2/N2气吸附30 min, 然后切换N2气吹扫, 待仪器基线平稳后, 以10 ℃/min的速率升温进行测试, 采用热导检测器检测SO2的脱附信号.
图1示出了CeTiOx催化剂的NH3-SCR反应活性和N2选择性. 由图1(A)可见, 在所有催化剂上NO转化率均随着反应温度的升高而明显提高, 在550 ℃时略有下降. 这可能是由于在较高温度下, 部分NH3被催化氧化为NOx, 从而降低了催化剂的脱硝性能[16]. CeTiOx-A催化剂表现出了最优异的NH3-SCR反应活性, 其NO转化率在280 ℃时即达到93%, 并且具有最宽的T90温度窗口(280~550 ℃). CeTiOx-B催化剂的NO转化率在320 ℃时达到94%, T90温度窗口为320~550 ℃. 这表明以Ce2(C2O4)3为前驱体可得到NH3-SCR反应活性较高的CeTiOx催化剂. 在经过高硫气氛SCR反应后, 40CeTiOx-A催化剂和60CeTiOx-A催化剂的NO转化率基本无变化. 40CeTiOx-B催化剂与40CeTiOx-A及60CeTiOx-A催化剂相比, 在320 ℃以下的NO转化率略低, 320 ℃以上基本相同, 但总体的NO转化率明显高于60CeTiOx-B. 若延长CeTiOx-B催化剂的反应时间, NO转化率可能会进一步降低. 由图1(B)可见, 各催化剂的N2选择性随着反应温度的升高而逐渐提高, 在活性温度窗口内的N2选择性均保持在99%以上, 在550 ℃时N2选择性略有下降, 这可能与NH3被催化氧化为NOx有关. CeTiOx-A催化剂的N2选择性最佳, 在280 ℃时即达到99%. 以上结果表明, 以Ce2(C2O4)3为前驱体得到的CeTiOx催化剂具有更高的的N2选择性.
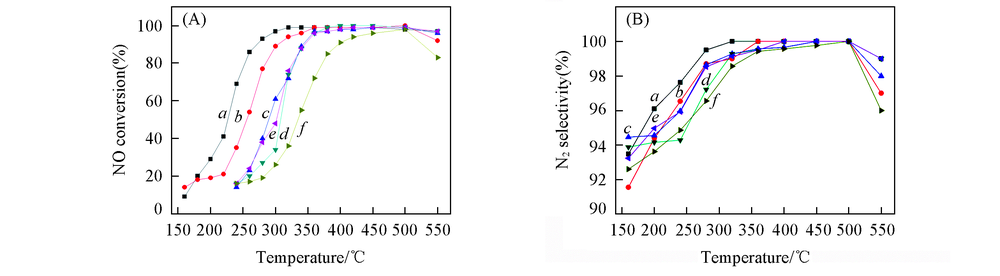 | Fig.1 NH3-SCR activity(A) and N2 selectivity(B) over the catalysts Reaction conditions: 0.1%NO+0.1%NH3+6%O2+0.0175%SO2+6%H2O, He balance, GHSV: 3× 104 h-1. a. CeTiOx-A; b. CeTiOx-B; c. 40CeTiOx-A; d. 40CeTiOx-B; e. 60CeTiOx-A; f. 60CeTiOx-B. |
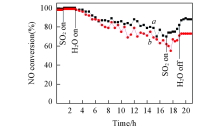
SO2和H2O会影响催化剂的NH3-SCR反应活性. 考察了300 ℃下SO2和H2O对CeTiOx催化剂的NH3-SCR反应活性的影响. 图2示出CeTiOx-A和CeTiOx-B催化剂在SO2和H2O存在时的实验结果. 可见, 当向反应系统通入体积分数为0.0175%的SO2后, 2个催化剂的NH3-SCR反应活性均有小幅度提升. 这可能是由于通入的少量SO2吸附在催化剂表面, 增强了催化剂的表面酸性所致[17]. 将体积分数为6%的H2O引入反应体系后, CeTiOx-B催化剂的NO转化率下降更明显, 反应14 h后降至62%, 而CeTiOx-A催化剂的NO转化率在反应14 h后仍在70%以上. 当切断SO2后, CeTiOx-A和CeTiOx-B催化剂的NO转化率分别恢复至82%和71%; 再将H2O切断后, 二者的NO转化率分别恢复至88%和73%. 上述结果表明, 以Ce2(C2O4)3为前驱体的CeTiOx催化剂表现出较高的抗硫抗水性能.
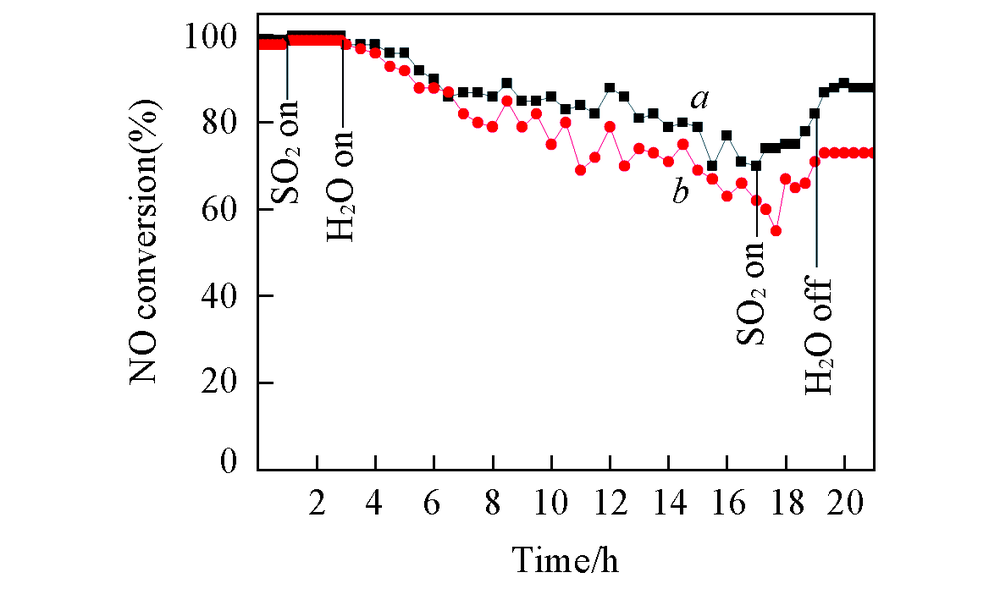 | Fig.2 NH3-SCR activity over CeTiOx-A(a) and CeTiOx-B(b) in the presence of SO2/H2O at 300 ℃ Reaction condition: 0.1%NO+0.1%NH3+6%O2+0.0175%SO2+6%H2O, N2 balance, GHSV: 3× 104 h-1. |
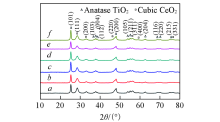
图3为CeTiOx催化剂的XRD谱图. 可见, 无论是新鲜的CeTiOx催化剂还是反应后的CeTiOx催化剂, 均在2θ =25.2° , 36.8° , 37.8° , 38.5° , 47.9° , 53.7° , 55.0° , 62.6° , 68.7° , 70.4° 和75.0° 处出现了特征衍射峰, 分别归属为锐钛矿相结构TiO2(JCPDF: 21-1272)的(101), (103), (004), (112), (200), (105), (211), (204), (116), (220)和(215)晶面. 在2θ =28.5° , 33.1° , 47.3° , 56.2° , 69.2 ° 和76.5° 处出现特征衍射峰, 分别归属为立方萤石相结构CeO2(JCPDF: 34-0394)的(111), (200), (220), (311), (400)和(331)晶面. 与新鲜的CeTiOx催化剂相比, 在含有0.15%的SO2的反应气氛下, 在分别反应40和60 h的CeTiOx催化剂上, 并未发现有关硫酸盐物种的特征衍射峰. 这可能是因为硫酸盐物种以无定形的状态存在, 或者形成的硫酸盐物种太少, 以至于无法观察到[18, 19]. 不同Ce前驱体制备的CeTiOx系列催化剂的晶相结构并无太大差别, 表明催化剂的晶相结构并不是影响其NH3-SCR反应活性差别的主要因素.
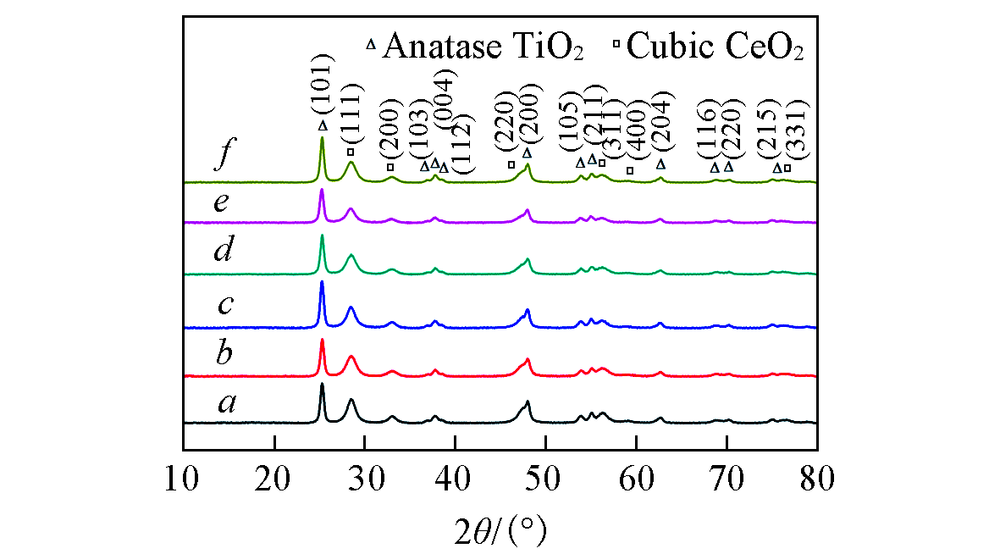 | Fig.3 XRD patterns of CeTiOx-A(a), CeTiOx-B(b), 40CeTiOx-A(c), 40CeTiOx-B(d), 60CeTiOx-A(e) and 60CeTiOx-B(f) |
表1给出了CeTiOx催化剂样品的元素组成、 比表面积和孔容数据. 由表1可见, 各催化剂中TiO2和CeO2的实际质量比与投料质量比相近. 反应后的CeTiOx催化剂的SO3含量比反应前明显增大, 表明反应后生成了部分硫酸盐物种. 结合XRD表征结果可知, 这些硫酸盐物种可能以无定形的状态存在于催化剂中. 通过比较各催化剂的比表面积可以发现, 反应后的催化剂比新鲜催化剂的比表面积显著减小. 这可能是由于反应生成的硫酸盐物种沉积在催化剂的表面, 堵塞了孔道[20]. 同时, 还发现与CeTiOx-B系列催化剂相比, CeTiOx-A系列催化剂具有更大的比表面积和孔容, 有助于催化剂上气体的吸附和脱附. 这可能是CeTiOx-A系列催化剂具有较高的NH3-SCR反应活性的原因之一.
| Table 1 Component content, BET surface area and pore volume of the catalysts |
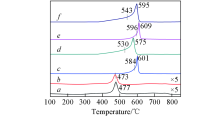
利用H2-TPR实验对CeTiOx催化剂进行了氧化还原性能分析, 结果如图4所示. 可见, 新鲜的CeTiOx-A和CeTiOx-B催化剂分别在477和473 ℃出现还原峰, 归属于分散在TiO2表面上Ce4+的还原. 由表2可知, 单位质量的新鲜催化剂的总耗氢量顺序为CeTiOx-A> CeTiOx-B, 表明CeTiOx-A催化剂具有较高的活性氧浓度, 因而CeTiOx-A催化剂的NH3-SCR反应活性高于CeTiOx-B催化剂. 反应后的CeTiOx催化剂在570~610 ℃范围内出现了较强的还原峰, 可归属于硫酸盐物种和次表面或者更深层的CeO2的还原[21, 22, 23]. 根据文献[24]报道, 硫酸盐弱吸附在TiO2上, 在约300 ℃可完全解吸, 当SCR反应进行一定时间后, 在CeTiOx催化剂上生成了金属硫酸盐Ce(SO4)2和Ce2(SO4)3物种[23]. 在所制备的催化剂上, 这些硫酸盐物种的还原峰温度顺序为60CeTiOx-A> 40CeTiOx-A> 60CeTiOx-B> 40CeTiOx-B, 这是因为硫酸盐物种可能以不同的形态存在于催化剂表面. 由表2可知, 催化剂的耗氢量顺序为60CeTiOx-B> 40CeTiOx-B> 60CeTiOx-A> 40CeTiOx-A, 表明CeTiOx-A系列催化剂比CeTiOx-B系列催化剂更容易生成较难还原的硫酸盐物种.
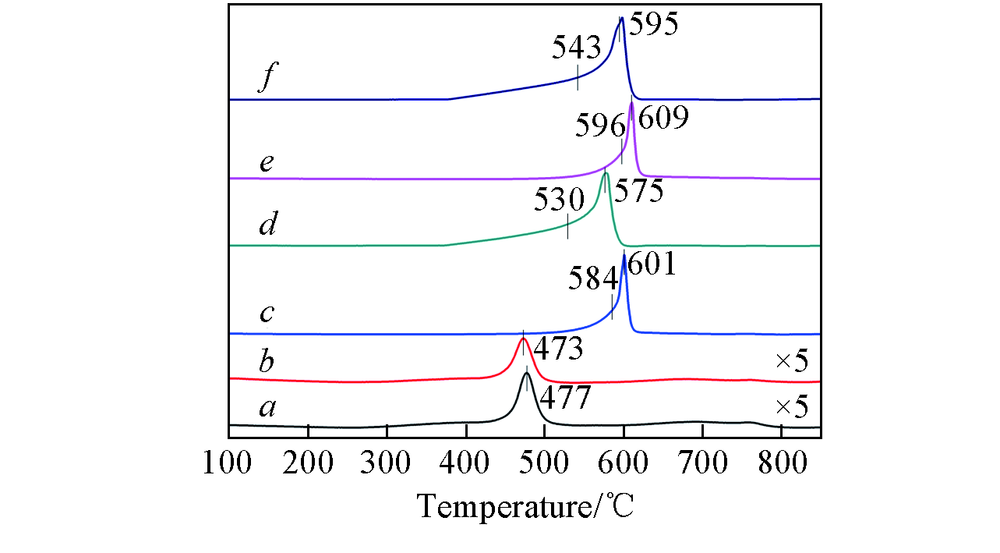 | Fig.4 H2-TPR profiles of CeTiOx-A(a), CeTiOx-B(b), 40CeTiOx-A(c), 40CeTiOx-B(d), 60CeTiOx-A(e) and 60CeTiOx-B(f) |
| Table 2 H2 consumption of the catalysts |
图5(A)为CeTiOx催化剂的T
CeO2+SO2+O2→Ce2(SO4)3 (3)
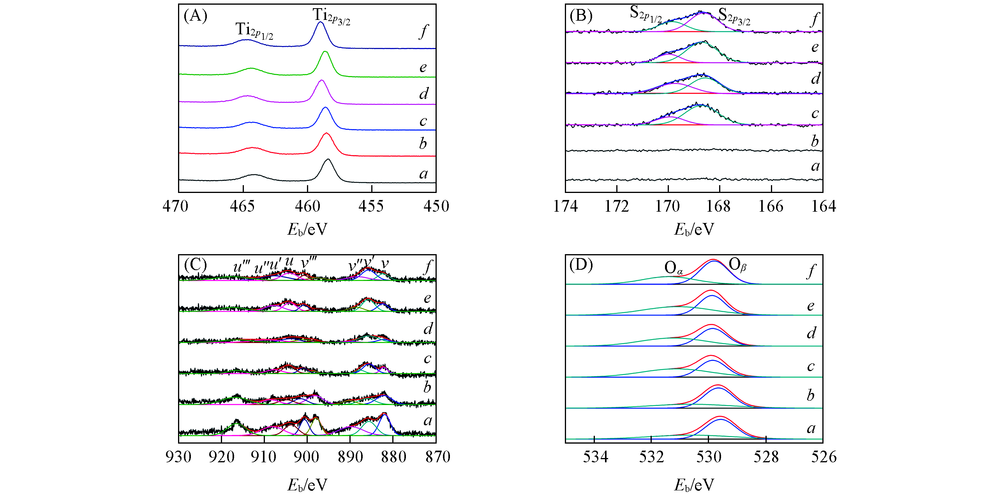 | Fig.5 XPS results of T a. CeTiOx-A; b. CeTiOx-B; c. 40CeTiOx-A; d. 40CeTiOx-B; e. 60CeTiOx-A; f. 60CeTiOx-B. |
| Table 3 XPS results of the catalysts |
催化剂表面的Ce3+会导致电荷不平衡, 产生大量的不饱和化学键, 使催化剂具有更好的氧化还原性能, 有利于提高催化剂的NH3-SCR反应活性[25, 32]. 图5(D)为催化剂的
研究表明, 催化剂的表面酸性对NH3-SCR反应活性具有重要的影响[34, 35]. 图6(A)示出了不同Ce前驱体制备的CeTiOx催化剂的NH3-TPD实验结果. 可见, 每个样品均具有2个较强的脱附峰, 在100~270 ℃范围内NH3的脱附可归属于Brö nsted酸性位上弱吸附NH3的脱附, 在270~540 ℃范围内NH3的脱附可归属于Lewis酸性位上强吸附NH3的脱附[33]. 反应后CeTiOx催化剂的总酸量比新鲜催化剂明显增大, 表明表面硫酸盐物种可以提供酸性位[36]. 反应后的CeTiOx-A系列催化剂的总酸量远大于反应后的CeTiOx-B系列催化剂, 表明CeTiOx-A系列催化剂在反应后生成了较多的表面硫酸盐物种, 与XPS表征结果一致. CeTiOx-A系列催化剂与CeTiOx-B系列催化剂相比, 无论是新鲜还是反应后的样品, 均具有较大的脱附峰面积, 即具有较大的总酸量. 同时, CeTiOx-A系列催化剂还具有较多的强Lewis酸性位数量, 催化剂表面的酸性位可以吸附较多的NH3, 有利于NO转化率的提高[37]. 这可能是CeTiOx-A系列催化剂具有较高的NH3-SCR反应活性的重要原因.
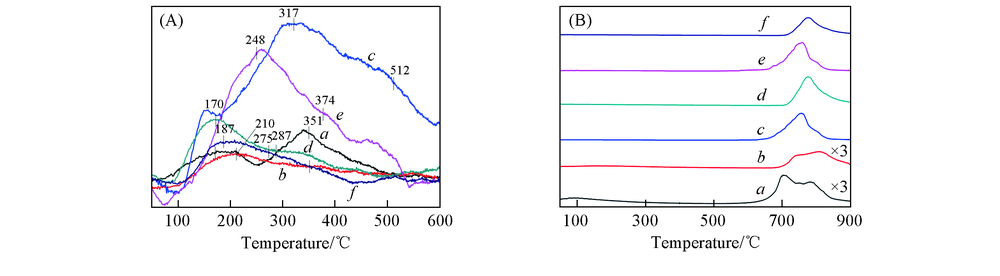 | Fig.6 NH3-TPD(A) and SO2-TPD(B) profiles of CeTiOx-A(a), CeTiOx-B(b), 40CeTiOx-A(c), 40CeTiOx-B(d), 60CeTiOx-A(e) and 60CeTiOx-B(f) |
为了考察CeTiOx催化剂对SO2的吸附情况, 进行了SO2-TPD实验. 如图6(B)所示, 在600~900 ℃范围内, 每个样品均出现了较强的脱附峰, 可归属为催化剂上表面和体相硫酸盐的分解. 无论是新鲜还是反应后的CeTiOx-A系列催化剂, SO2在其上的脱附量均大于CeTiOx-B系列催化剂, 前者也具有较低的SO2脱附温度, 说明CeTiOx-A系列催化剂生成了更多易分解的硫酸盐物种.
分别以Ce2(C2O4)3和Ce(SO4)2为Ce前驱体, 利用固相球磨法制备了系列CeTiOx催化剂, 比较了2种Ce前驱体制备的CeTiOx催化剂的NH3-SCR反应活性, 发现CeTiOx-A[以Ce2(C2O4)3为前驱体]催化剂比CeTiOx-B[以Ce(SO4)2为前驱体]催化剂具有更高的NH3-SCR反应活性和更好的抗硫抗水性能, 在280~550 ℃范围内, NO转化率能保持在90%以上. CeTiOx-A催化剂比CeTiOx-B催化剂具有更大的比表面积和孔容, 有助于反应物分子在催化剂表面的吸附. CeTiOx催化剂表面的Ce3+和吸附氧(Oα )物种有助于NO物种的吸附和活化, 较多的强Lewis酸性位可以吸附活化的NH3分子, 有利于催化剂上NH3-SCR反应的进行. 考察了CeTiOx催化剂在含有高浓度SO2的反应气氛下反应后催化剂的NH3-SCR反应活性, 结果显示, 反应后CeTiOx催化剂比新鲜CeTiOx催化剂具有较窄的反应活性温度窗口. 其原因是在CeTiOx催化剂表面生成了硫酸盐物种, 堵塞了催化剂表面的活性位点, 降低了催化剂的NH3-SCR反应活性.
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|
| [20] |
|
| [21] |
|
| [22] |
|
| [23] |
|
| [24] |
|
| [25] |
|
| [26] |
|
| [27] |
|
| [28] |
|
| [29] |
|
| [30] |
|
| [31] |
|
| [32] |
|
| [33] |
|
| [34] |
|
| [35] |
|
| [36] |
|
| [37] |
|