




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
C ε3-C ε4蛋白寡核苷酸适配子结合位点的预测与筛选
引用本文
刘中成, 刘世芳, 张苏, 杨艳蕾, 李飞, 张楠, 袁欣, 张艳芬. C ε3-C ε4蛋白寡核苷酸适配子结合位点的预测与筛选 [J]. 高等学校化学学报, 2019,40(1): 83-89.
LIU Zhongcheng, LIU Shifang, ZHANG Su, YANG Yanlei, LI Fei, ZHANG Nan, YUAN Xin, ZHANG Yanfen. Structure Prediction and Screening of Oligonucleotide Aptamers Target C ε3-C ε4 Protein† [J]. Chemical Journal of Chinese Universities, 2019,40(1): 83-89.
Doi:10.7503/cjcu20180428
Permissions
LIU Zhongcheng, LIU Shifang, ZHANG Su, YANG Yanlei, LI Fei, ZHANG Nan, YUAN Xin, ZHANG Yanfen. Structure Prediction and Screening of Oligonucleotide Aptamers Target C ε3-C ε4 Protein† [J]. Chemical Journal of Chinese Universities, 2019,40(1): 83-89.
Doi:10.7503/cjcu20180428
Copyright©2019, 《高等学校化学学报》编辑部
版权所有
C ε3-C ε4蛋白寡核苷酸适配子结合位点的预测与筛选
联系人简介: 张艳芬, 女, 副教授, 主要从事微生物生化药学方面的研究. E-mail: zhangjing@hbu.edu.cn
摘要
采用NPDock程序对C ε3-C ε4蛋白与其核酸适配子A1的结合位点进行了预测与筛选, 筛选出A1与C ε3-C ε4蛋白结合的关键位点. 同时, 根据蛋白与DNA片段复合物结合界面中氨基酸残基和碱基统计分析发现, 结合界面氨基酸富集碱基G能力最强, 富集碱基T和C能力次之. 本文建立了以NPDock程序虚拟对接为基础的高效适配子优化方法, 为相关研究提供了实验参考.
关键词:
C ε3-C ε4蛋白;; NPDock程序; 分子对接; 适配子筛选
中图分类号:O621
文献标志码:A
Structure Prediction and Screening of Oligonucleotide Aptamers Target C ε3-C ε4 Protein†
LIU Zhongcheng1, 2 , LIU Shifang1, 2 , ZHANG Su1, 2 , YANG Yanlei1, 2 , LI Fei1, 2 , ZHANG Nan1, 2 , YUAN Xin1, 2 , ZHANG Yanfen2, 3, * 
Abstract
The prediction and selection of binding sites between aptamers and targets are the key steps in SELEX process, however, the conventional screening process is complicated. As a new convenient screening method, the virtual screening based on computer has widely used in aptamers screening. The nucleic acid-protein dock(NPDock) program was used to predict and screen the binding site of C ε3-C ε4 protein and its aptamer A1 based on their molecular docking, and the key site of binding to C ε3-C ε4 protein was screened. According to the amino acid residues and bases in the binding interface between the protein and the DNA complexes by virtual docking, the results showed that the amino acid in the binding interface is most capable of enriching the base G, followed by the ability of enriching the base T and the base C. An efficient optimization method was established based on NPDock docking, which would provide an experimental reference for related researches.
Keyword:
C ε3-C ε4 protein ;; Nucleic acid-protein dock program; Molecular docking; Aptamers screening
适配子是指经指数富集的配体系统进化技术(SELEX)筛选的能特异性与靶点结合的单链DNA或RNA, 具有亲和性高、 特异性强、 靶标范围广、 分子量小、 无批次间差异、 易于修饰与合成、 稳定性好、 便于运输和储存及无免疫原性等优势. 在基础研究、 临床诊断与治疗[1]、 生化分析及新药研发等领域展示了广阔的应用前景[2]. 经初步筛选获得的核酸适配子作为先导分子, 通常需要进行结构优化, 寻找与靶物质结合的关键位点, 明确最优结合碱基后进行化学修饰才能更好地发挥治疗作用. 预测适配子与靶物质作用结构, 筛选关键结合位点是适配子缩减、 优化筛选及后期药物开发的必要环节. 目前, 基于计算机技术进行分子对接常被用于研究药物与靶点之间的相互作用以及虚拟筛选具有生物活性的小分子[3]. 用于分析蛋白与核酸相互作用的对接程序主要有Automated dock(AutoDock)[4], High Ambiguity Driven Protein-protein Docking(HADDOCK)[5], Patch-Dock[6], Zhiping DOCK(ZDOCK)[7]和Hex Protein Docking(HEX)[8]等, 这些程序均以相关分子力学方法为基础用于探寻DNA分子与蛋白质靶点间的结合模式. 虽然, 其中一些方法考虑了配体分子的柔性, 将参与对接过程的DNA构象视为发生变化, 并且通过改变分子的空间位置与姿态进行对接; 但是, 其通过模拟复合物对接构象, 经聚类分析选择最佳相互作用的构象, 适用于生物大分子间相互作用模拟. 核酸的结构功能特异, 具有高度电荷, 适配子分子较小, 对接过程中构象存在较大变化, 故应用以上对接方法具有一定的局限性[9, 10].
Nucleic acid-Protein Dock(NPDock)是一种针对蛋白质-核酸对接开发的对接方法[11], 它通过使用特定的蛋白质-核酸统计势对模拟的复合物进行评分和选择, 实现了一种独特的工作流, 包括GRAMM程序, 用于粗粒表示的蛋白质-RNA复合物评分的DARS-RNP和QUASI-RNP统计势能[12], 以及用于对蛋白质-DNA复合物评分的QUASI-DNP, DFIRE与Varani组合程序. 前文[13]利用SELEX技术筛选得到Cε 3-Cε 4蛋白核酸适配子A1, 可抑制由IgE介导的过敏反应. 为了获得适配子与靶蛋白的结合位点, 便于后期适配子修饰及化学合成, 本文利用NPDock方法将适配子A1与Cε 3-Cε 4蛋白进行对接, 明确了适配子与靶蛋白结合的关键位点, 为开发适配子A1药物提供了实验基础, 同时为适配子筛选与开发提供参考.
1 实验部分
1.1 实验材料及软件
Cε 3-Cε 4蛋白特异性适配子A1的核酸序列为ACCGACCGTGCTGGACTCTGCCCCGCTGCCTTGCATCTGTCTTGTCTTTAATGGTATCCACAGTATGAGCGAGCGTTGCG. 利用布鲁克海文蛋白质数据库(Brookhaven Protein Data Bank)检索Cε 3-Cε 4蛋白的晶体结构; 以大分子构建软件(Macromolecule Builder, MMB)预测适配子结构; 以在线预测软件NPDock(http://genesilico.pl/NPDock/submit)对适配子与靶蛋白进行分子对接; 以PyMol程序(https://pymol.org/2/)分析对接获得的DNA-蛋白质复合物的结构.
1.2 配体的准备
实验选用靶向Cε 3-Cε 4的特异性适配子A1, 由80个碱基构成的一段DNA序列, 两端分别为20 bp的固定序列, 中间为40 bp的随机序列. 根据其一级结构, 按照碱基互补配对原则将其截取为发夹结构序列, 根据上述信息构建MMB参数文件, 在命令提示符中生成DNA片段的结构文件, 文件格式为pdb.
MMB的参数设置如下: 基础相互作用比例因子200; 报告间隔4.0; 温度10.0; 数据报告间隔10; 随机初始速度TRUE.
1.3 蛋白受体的准备
PDB数据库是目前最主要的蛋白质分子数据库, 目前共收录了7万余种晶体. 通过检索晶体蛋白的PDB号(1FP5), 在PDB蛋白库中搜索Cε 3-Cε 4结构, 找到PDB文件并下载, 用PyMol显示其结构为Cartoon形式(见图1).
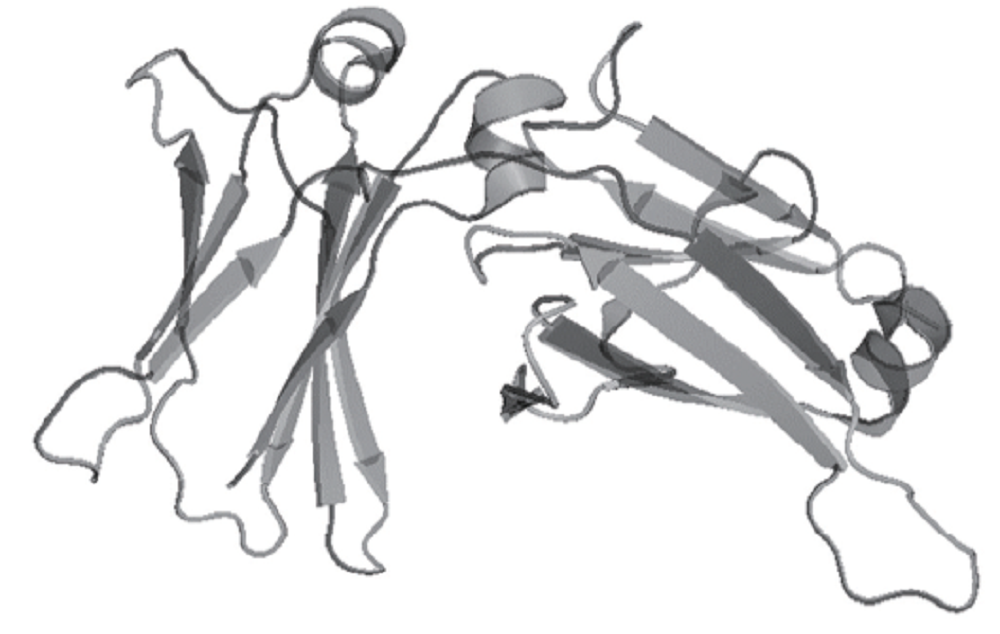 | Fig.1 3D structure of Cε 3-Cε 4 protein |
1.4 NPDock分子对接
以MMB产生的46种DNA短发夹的三维结构为配体, 以靶点蛋白Cε 3-Cε 4(PDB代码1FP5)为受体, 使用NPDock程序搜索配体与受体相互作用的构象[11] . 设置NPDock相关参数, 进行对接. 在每次对接中生成的配体与靶点蛋白复合结构模型中, 根据生成的最优势构象进行统计分析, 选出打分最优且构象合理的结果.
NPDock对接的参数具体为预产生诱饵: 20000, 聚类最佳得分模型: 100, 聚类均方根偏差中断: 0.5 nm, 模拟步骤: 1000, 第一步与最后一步模拟温度: 15000和295 K.
1.5 蛋白质与DNA适配体复合物的结构分析
利用PyMol软件对对接获得的DNA-蛋白质复合物的结构进行分析, 统计结合界面的氨基酸残基, 用于分析蛋白与DNA片段对接时氨基酸的偏好性; 并对各个氨基酸与4个碱基之间作用频率进行统计分析, 找出各个氨基酸富集的碱基. 为了发现能与靶点蛋白有较强作用的DNA片段, 对短发夹DNA结构与蛋白作用的碱基进行统计分析, 基于常规结构生物学知识, 对DNA适配子A1与Cε 3-Cε 4蛋白关键结合位点进行详细分析, 在此基础上预测结合能力较强的DNA短发夹结构.
2 结果与讨论
2.1 DNA适配体及蛋白Cε 3-Cε 4的结构
以DNA适配子A1的一级序列为基础, 按照碱基互补配对原则, 对配对碱基形成的发夹结构依次进行截取, 获得了48个DNA片段(见表1), 其中序列18, 25和26是由3对碱基对形成的发夹结构, 其余序列均由2对碱基对形成.
| Table 1 Sequences for DNA aptamers |
使用MMB程序, 设置碱基对序列和温度等参数, 构建了46个PDB格式的DNA适配子发夹结构, 其中序列2和序列24的发夹结构构建失败. 这46个适配子发夹结构长度各不相等, 短至5个碱基(序列5), 长则可达26个碱基(序列14), 但大部分均以短序列存在, 以获取与蛋白作用的最短核心部位. 利用PyMol软件将短发夹适配体的结构显示成Cartoon, 图2显示了长度各不相同的短发夹适配体结构, 序列4, 18和21[图2(A), (C)和(D)]为分别由6, 9和11个碱基形成的较短发夹结构; 序列10, 22和29[图2(B), (E)和(F)] 为分别由24, 18和18个碱基, 通过2个碱基互补配对形成的1个大环状发夹结构. 构建适配子3D结构是蛋白与核酸对接的关键[14], 利用RNA composer程序[15]对适配子A1的发卡结构进行了预测[图2(G)~(L)], 通过对比发现2种方法具有一定的相似性, MMB比RNA Composer更具灵活性, 可以指定碱基配对等参数, 允许分子的不同区域以不同的灵活性进行处理, 能够人为地控制核酸的空间结构[16, 17].
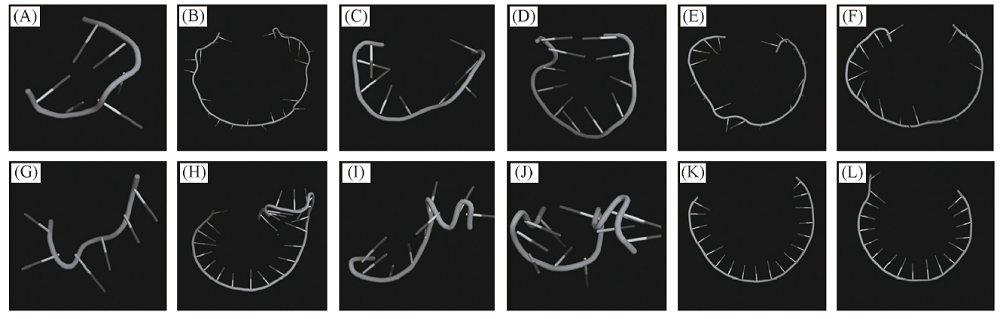 | Fig.2 3D structures prediction of DNA short hairpin (A)— (F) 3D structures of sequences 4, 10, 18, 21, 22, 29 by MMB; (G)— (L) 3D structures of sequences 4, 10, 18, 21, 22, 29 by RNA composer. |
2.2 NPDock对接结果
NPDock软件简洁易用, 界面友好, 可以同时进行多个对接, 以邮件方式接收结果[18, 19]. 使用在线预测软件NPDock, 对每个对接任务生成的20000个蛋白与DNA适配体的复合物结构进行构象分析, 分别选择打分较高的构象, 最终得到了46个DNA-蛋白复合物的优势构象结构, 复合物结构是由网站生成的JSmol三维可视化的复合物结构(见图3).
 | Fig.3 3D conformation of DNA short hairpins and Cε 3-Cε 4 protein interaction predicted by NPDock (A)— (C) 3D visualization of the 4, 18, 21 best scored complex. |
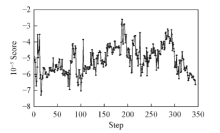
为了查看对接过程中每一步DNA片段与蛋白结合模式打分的区别, 绘制了序列8片段与靶蛋白对接过程打分变化图(图4), Step数为1~350. 由图4可见, 随着Step数目增多, 打分变化较大, 说明对接模拟过程对DNA片段与靶蛋白结合构象进行了充分搜索, 这为获取能量最低(即打分最好的构象)提供了前提条件. 本文比较了ZDOCK与NPDOCK的对接结果, 发现二者具有明显差异(图S1和表S1, 见本文支持信息).
 | Fig.4 Plot illustrating score changes during a simulation |
为了找到与靶蛋白Cε 3-Cε 4结合能力最强的DNA片段, 对获得的短发夹适配体与蛋白复合物构象的打分进行了统计, 结果如图5所示, 分值较高的DNA片段主要集中于2个部分: 序列6~14和20~32, 表明这2部分序列中可能含有DNA适配子与蛋白发生亲和作用的区域.
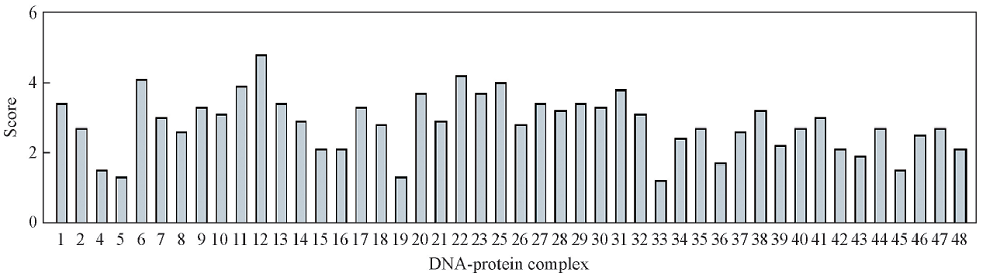 | Fig.5 Scoring statistic of DNA fragments and Cε 3-Cε 4 protein docking results |
2.3 蛋白质与DNA适配体复合物的结构分析
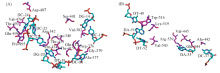
根据构象的打分结果, 从中筛选出8组优势构象(见表2)作为与蛋白对接的关键区域, 并对其进行分析(图6). 由图6(A)可见, 序列11与蛋白相互作用的结合界面上有9种氨基酸与DNA片段上的碱基发生相互作用, 其中Thr出现频率最高, 在序列11与蛋白作用的复合物结构中氨基酸Thr出现4次与碱基产生相互作用, 分别为Thr415-T19, Thr433-G20, Thr434-G20和Thr493-C24. Thr是极性氨基酸, 侧链上带有1个能够与DNA磷酸骨架形成氢键的— OH基团, 且Thr为极性氨基酸, 溶于水, 易暴露于蛋白质分子表面, 比其它氨基酸具有较强的偏向性. 由图6(B)可见, 序列22与蛋白相互作用中有5种氨基酸残基对DNA片段与蛋白复合物结构的相互作用有贡献, 其中Arg和Lys均参与2次, 分别为Arg539-A35, Arg513-T49和Lys497-G39, Lys519-A51. Arg和Lys为碱性氨基酸, 可与DNA片段发生正负电荷间的极性作用.
| Table 2 Best sequences of DNA aptamers from docking results |
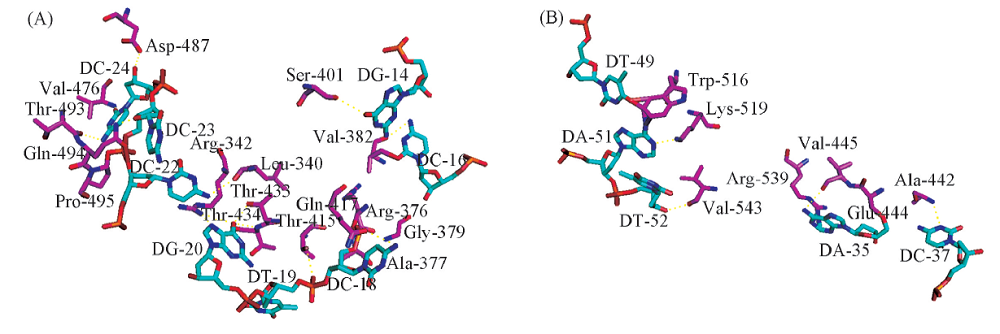 | Fig.6 Predictions for protein-DNA complexes (A) The site of interaction of sequence 11 protein complex, the purple are the amino acid residues Thr, Ser, Asp, Lys and His. The cyan are the base T, C and G. (B) The site of interaction of sequence 22-protein complex, the purple are the amino acid residues Ala, Arg, Val, Lys, Glu and Arg. The cyan are the base A, T, C and G. |
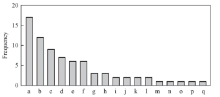
为进一步了解蛋白与DNA片段之间的相互作用, 对8组DNA片段与蛋白复合物结构相互作用的氨基酸及其使用频率进行统计, 找出蛋白与DNA片段相互作用时氨基酸的偏好性. 如图7所示, 在76个作用位点中, 有17个为Thr(苯丙氨酸), 占最大比重; 占比重较大的氨基酸还有Arg, Ser, Gln和Lys等; 占比重较小的是His, Ile, Leu, Met和Tyr, 且仅出现1次.
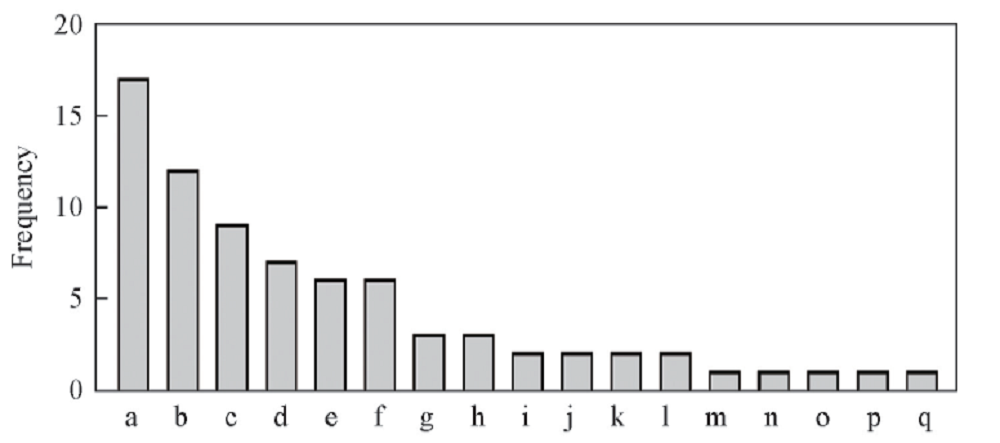 | Fig.7 Frequency of amino acid usage at the site of complex interaction a. Thr; b.Arg; c. Ser; d. Gln; e. Lys; f. Val; g. Asp; h. Pro; i. Glu; j. Phe; k. Ala; l. Asn; m. His; n. Ile; o. Leu; p. Met; q. Tyr. |
蛋白与DNA的相互作用由氨基酸与碱基间的作用力决定, 以序列22-蛋白复合物为例进行分析, 在DNA片段与蛋白复合物的结合界面上共有8组氨基酸与碱基, 主要表现为氢键和范德华作用2种作用力. 图8为4组氨基酸与碱基发生氢键作用的界面, Ala与碱基C是由氨基酸上的— NH2与碱基C嘧啶环上的N原子间形成氢键[图8(A)]; Val与碱基A是由氨基酸上的O原子与碱基A嘌呤环上的— NH2形成氢键[图8(B)]; Lys与碱基A是由氨基酸上的— NH2与碱基A嘌呤环上的N原子形成氢键[图8(C)]; Val与碱基T是由氨基酸的— OH基团与碱基上的O原子形成氢键[图8(D)]. 可见, 碱基A和T结构中嘌呤环上的N原子以及— NH与氨基酸易发生氢键作用; 碱基C和G结构中嘧啶环上的N原子也容易与含有— OH和— NH的氨基酸发生氢键作用. Arg和Lys等碱性氨基酸的侧链上含有胍基、 氨基和咪唑基这类碱性基团, 使结合界面表现出大量的正电荷, 从而与DNA分子的磷酸基团带有的大量负电荷形成静电作用[20].
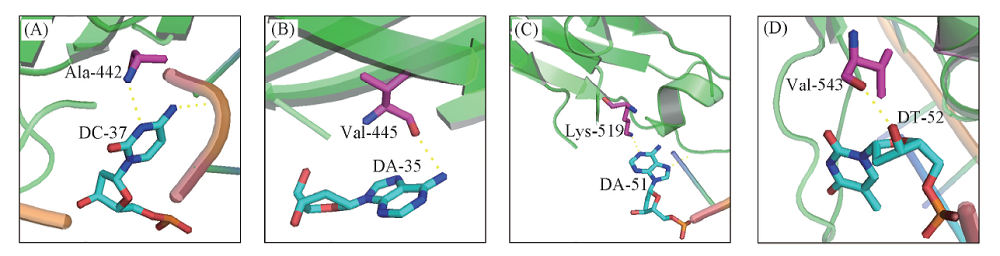 | Fig.8 Binding patterns between the amino acid-nucleotide pairs |
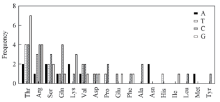
对DNA片段与氨基酸结合界面的各个氨基酸与4个碱基间相互作用的频率进行了统计, 如图9所示, Thr与碱基G的作用能力明显强于其它氨基酸, Arg次之; Thr, Arg和Gln与碱基C均有较强的结合能力; Thr与碱基T间的结合力明显高于其它氨基酸, Arg, Ser和Val与碱基T也有较高的结合作用; 而碱基A在复合物作用界面中与氨基酸结合能力则比其它碱基弱.
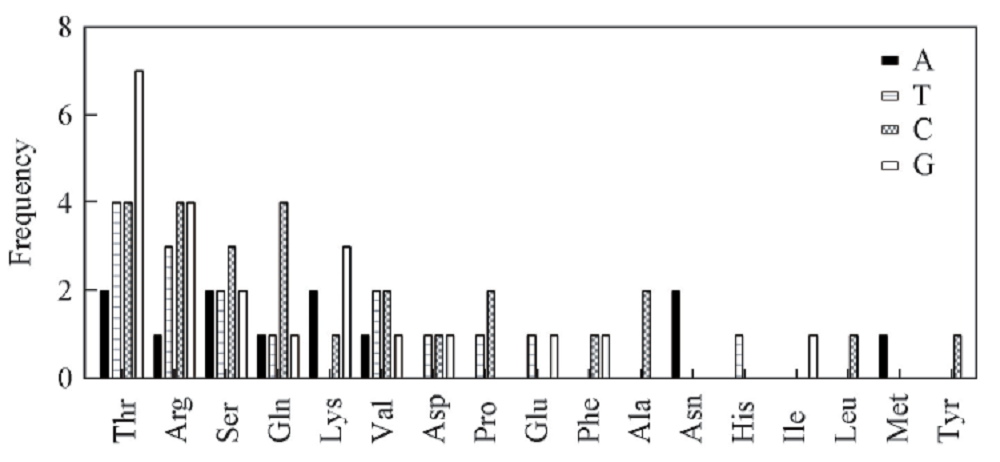 | Fig.9 Distribution of the frequency of the interaction between each of the amino acids and each of the four nucleotides |
对46个DNA片段-蛋白复合物中作用位点的碱基进行统计分析发现, 碱基G与蛋白的结合率为30.16%, 高于其它碱基, 而碱基A与蛋白的作用最低(14.88%), 碱基T(28.1%)和C(26.86%)与蛋白作用的结合率大致相同. 此结果和蛋白与短发夹DNA复合物界面上氨基酸残基与碱基作用的规律一致, 说明一段序列中若碱基G, C和T占序列的百分含量高, 则这段序列与蛋白的结合率较高, 也解释了序列11与蛋白结合打分高的原因. 由表2可见, 序列4, 10和11中包含1段相同的序列GGACTC, 说明与蛋白结合时这段序列对蛋白的亲和作用较强. 而TGTCTTGTCTTTAA这段序列在序列18, 21, 22, 23和29中均出现, 其对适配子与蛋白结合起着非常重要的作用. 因此将蛋白Cε 3-Cε 4与核酸发挥作用的关键区域定位于GGACTC和TGTCTTGTCTTTAA这2段序列中, 但GGACTC片段是所有适配子均含有的固定序列的一部分, 因此排除GGACTC与蛋白Cε 3-Cε 4的结合作用, 推测TGTCTTGTCTTTAA可能是与蛋白Cε 3-Cε 4作用的关键结合位点. 赵丽健[21]对适配子A1与Cε 3-Cε 4结合关键位点进行了筛选, 得到GGGGTCTTGTCTTTAACCCC序列是蛋白Cε 3-Cε 4的亲和区域, 通过体内、 体外实验结果证明TGTCTTGTCTTTAA为蛋白Cε 3-Cε 4与适配子A1的作用位点. 预测结果与实验结果十分吻合, 证明该方法可行、 有效.
3 结 论
首先通过MMB软件构建了适配子3D结构, 然后采用NPDock程序将Cε 3-Cε 4蛋白与其适配子A1对接, 并对对接复合物进行了分析, 筛选出与Cε 3-Cε 4蛋白结合的关键位点为TGTCTTGTCTTTAA序列. 通过统计并分析蛋白与DNA片段复合物结合界面中氨基酸残基和碱基, 得出结合界面氨基酸富集碱基G能力最强, 富集碱基T和碱基C能力次之. 本文对于理解蛋白质与DNA适配体相互作用的机制具有一定的意义, 也为适配子筛选提供了参考.
支持信息见http://www.cjcu.jlu.edu.cn/CN/10.7503/cjcu20180428.
参考文献
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|
| [20] |
|
| [21] |
|